1. 概述
普通的細菌毒素主要分成兩種,一種是細菌外毒素(Exotoxin):它是一種有毒性的蛋白質,是細菌在繁殖過程中所分泌在細菌體外產生的有毒物質。產生外毒素的菌株,大多是革蘭氏陽性菌,還有白喉桿菌、破傷風桿菌、肉毒桿菌、金黃色葡萄球菌以及少數革蘭氏陰性菌。另一種是細菌內毒素(Endotoxin,脂多糖):為革蘭氏陰性菌的細胞壁的產物,細菌在正常生活中時并不會自行產生,只有在細菌死亡自溶并附著在其它細胞上后,才顯示其毒性。
細菌內毒素與人類生活息息相關,人類賴以生存的水源中同樣含有細菌內毒素,其量約為1~100EU/ml。然而當細菌內毒素通過消化道進入人體時并不會產生影響,只有通過注射等方式進入血液時才會引起不同程度的危害。細菌內毒素少量進入血液后會被肝臟枯否細胞滅活,不會對機體造成損害,若大量進入血液后就會引起熱原反應,即發熱反應。因此,注射劑在產品研發上市過程中控制細菌內毒素很有必要。
2. 檢查方法
根據中國藥典2020版四部通則1143細菌內毒素檢查法中描述,細菌內毒素檢查包括兩種方法,即凝膠法和光度測定法,后者包括濁度法和顯色基質法。供試品檢測時,可使用其中任何一種方法進行試驗。當測定結果有爭議時,除另有規定外,以凝膠限度試驗結果為準。
目前,凝膠法檢查法使用的較多,是一種限度檢測或半定量的一種檢驗方法,光度測定法可定量檢測細菌內毒素。
2.1 凝膠法
2.1.1. 實驗原理
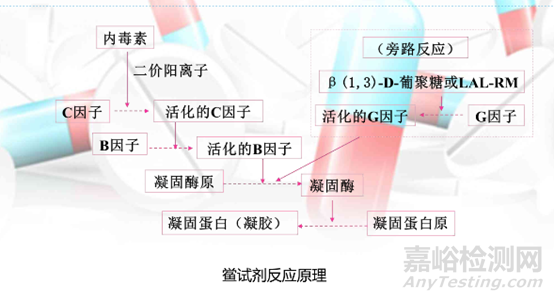
細菌內毒素的檢查原理是通過觀察鱟試劑與微量內毒素產生凝集反應的現象,來判斷供試品中細菌內毒素的限量是否符合規定。
鱟試劑是一種無菌冷凍干燥品,由海洋生物鱟的血液變形細胞溶解物制成,其中含有能被微量細菌內毒素和真菌葡聚糖激活的凝固酶原,凝固蛋白原,能夠檢測樣品中是否含有細菌內毒素和(1,3)-β-葡聚糖激,準確、快速地定性或定量。
反應原理如下:
2.1.2. 細菌內毒素限度的制定
一般情況下,是按照中國藥典2020版四部通則1143細菌內毒素檢查法進行計算:
3. 示例:XX注射液
3.1 限度制定依據
本品規格為300ml:API I 45g與API II 15g。參比制劑說明書中的常規用量用法為3~10min內滴注100ml,每1kg滴注7~20ml,每日API I最大劑量為200g,即本品每日最大劑量為1334ml。
根據上述公式,計算制劑的細菌內毒素限度L:L=K/M=5EU/(kg·h)/1334ml/(60 kg·h)=0.22EU/ml。
國家藥品審評中心化藥藥物評價《細菌內毒素檢查法研究中應注意的幾個方面》中指出,大輸液內毒素限度一般計為0.5EU/ml,所以擬定本品內毒素限度為0.5EU/ml。
3.2 物料控制
在注射劑中細菌內毒素貢獻占比最大,需要根據產品的細菌內毒素來計算控制原輔料的細菌內毒素。故需要對本品原輔包的細菌內毒素制定合理的來源控制和過程控制:
3.2.1. API I
根據中國藥典2020年版四部通則1143細菌內毒素檢查法中公式,計算 API I的細菌內毒素限度L:L=K/M=5EU/(kg·h) / 200g/(60 kg·h)=1.5EU/g
式中:
L為供試品細菌內毒素限值,以EU/mg表示;
K為人每千克體重每小時最大可接受的內毒素劑量,本品為注射劑,K=5EU/(kg·h);
M為人用每千克體重每小時的最大供試品劑量,以mg/(kg·h)表示。人均體重按60kg計算,本品中API I臨床每小時最大使用劑量為200g,故本品M=200g/(60kg·h)。
收緊細菌內毒素限度為≤0.5EU/g,并訂入API I進廠內控質量標準。
3.2.2. API II
本品規格為300ml:API I 45g與API II 15g。按照參比制劑說明書用法用量,每日API I最大劑量為200g,即本品每日最大劑量為1334ml,故每日API II最大劑量為66.67g。根據中國藥典2020年版四部通則1143細菌內毒素檢查法中公式,計算API II的細菌內毒素限度L:L=K/M=5EU/(kg·h) / 66.67g/(60 kg·h) = 4.5EU/g
收緊細菌內毒素限度為≤0.23EU/g,并訂入API II進廠內控質量標準。
3.2.3. 注射用水
參考ChP2020(注射用水細菌內毒素限度為≤0.25EU/ml),收緊限度為≤0.125EU/ml,并訂入內控質量標準。
3.2.4. 內包材
直接接觸產品內包材如西林瓶、膠塞、安瓿等,在生產使用時會要求除去細菌內毒素并進行驗證,驗證標準是內毒素能夠降低3個log值。在產品細菌內毒素貢獻占比較小。本品使用的內包材五層共擠輸液用膜袋(含塑料輸液容器接口和塑料輸液容器聚丙烯組合蓋)。參考FDA(≤0.5EU/ml),收緊包材組件的細菌內毒素限度為0.25EU/ml,并訂入五層共擠輸液用膜袋內控質量標準。
3.3 結論
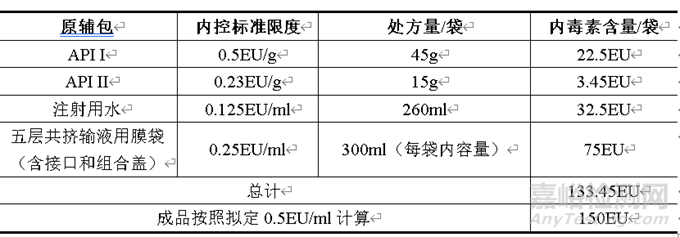
擬定本品的細菌內毒素限度為0.5EU/ml(即150EU/袋),原輔料、注射用水和內包材的細菌內毒素總量按照內控限度計算為133.45EU/袋,小于本品的擬定限度,計算結果具體見下表。從原輔料、注射用水和內包材方面可以有效控制本品的細菌內毒素,保證產品質量。
4. 實驗過程
4.1 確定最大有效稀釋倍數
確定供試品最大有效稀釋倍數(MVD)。
MVD是指在試驗中供試品溶液被允許達到稀釋的最大倍數,在不超過此稀釋倍數的濃度下進行內毒素限值的檢測。(如果無限稀釋供試品,內毒素可能會檢測不出來。)
MVD=cL/λ
式中:
L為供試品的細菌內毒素限值;
c為供試品溶液的濃度,當L以EU/ml表示時,則c等于1.0ml/ml,當L以EU/mg或EU/U表示時,c的單位需為mg/ml或U/ml。
λ為鱟試劑的標示靈敏度(EU/ml)【凝膠法】,或是標準曲線最低的內毒素濃度【光度測定法】。
4.2 鱟試劑的靈敏度復核試驗
鱟試劑的靈敏度(EU/ml)為在檢查法規定的條件下,使鱟試劑產生凝集的內毒素的最低濃度。
如果使用新批次的鱟試劑,或者試驗條件發生了改變時,包括任何可能影響檢驗結果的改變,都要進行鱟試劑靈敏度復核試驗。復核的目的是為了確認鱟試劑的靈敏度,同時也考察了檢驗人員操作方法,并確認了試驗條件是否符合規定。
4.3 干擾試驗
干擾試驗是為產品使用細菌內毒素檢查法提供依據,確定供試品在確定濃度下對內毒素和鱟試劑的反應都不存在干擾作用。在開發細菌內毒素的檢查方法時,試驗前須進行干擾試驗。當試驗條件發生了改變時,包括鱟試劑、供試品的配方、生產工藝改變或試驗環境中發生了任何變化時,須進行干擾試驗。
4.4 供試品檢查
使用已經過驗證的方法對制劑品種進行檢驗。
5. 小結
在注射劑的生產過程中,可能會從原輔料、生產過程、人員、設備以及環境等各個方面引入微生物及細菌內毒素。為了保證藥物使用的安全性,各國法規標準對于注射劑的細菌內毒素水平都有嚴格的規定,若沒有標準,也需要在研發過程中制定相應的內控標準。也同時要求生產企業在注射劑研發過程中采取合理的策略,嚴格控制注射劑中的細菌內毒素含量。