摘要: 污染控制是藥品生產(chǎn)質(zhì)量管理的核心內(nèi)容之一,該文基于藥品生產(chǎn)質(zhì)量管理的基本要求,對無菌藥品污染控制策略的要點進行了概述;通過對近年來國內(nèi)外無菌藥品檢查中在人員、設備與組件、設施環(huán)境、物料、工藝過程、檢測與預防等方面發(fā)現(xiàn)的污染控制的典型性及容易忽略的問題實例進行提煉與闡述,以期為無菌藥品生產(chǎn)企業(yè)在污染控制策略的設計與評估提供借鑒,為各類無菌藥品生產(chǎn)檢查提供參考。
藥品生產(chǎn)質(zhì)量管理規(guī)范(good manufacturing practice,GMP)旨在最大限度地降低藥品生產(chǎn)過程中污染、交叉污染以及混淆、差錯等風險。污染是藥品質(zhì)量最大的威脅,污染控制是藥品生產(chǎn)質(zhì)量管理的重要內(nèi)容,微生物、熱原和微粒等污染的控制更是無菌藥品生產(chǎn)質(zhì)量管理的核心要點,歷史上發(fā)生的多個藥害事件均涉及無菌藥品的污染[1]。鑒于無菌藥品污染控制的重要性及特殊性,國際上部分藥品檢查機構將藥品檢查員劃分為無菌藥品檢查員和非無菌藥品檢查員。近年來在國內(nèi)外藥品檢查中,針對無菌藥品污染控制方面發(fā)現(xiàn)的問題相對較多,部分問題對藥品質(zhì)量造成較大的潛在風險。考慮到上述原因,歐洲藥品監(jiān)督管理局(European Medicines Agency,EMA)新修訂的無菌藥品 GMP 附錄明確提出了污染控制策略的概念與要求[2],2022 年正式生效的國際 藥 品 檢 查 協(xié) 作 組 織 ( Pharmaceutical Inspection Co-operation Scheme,PIC/S) GMP 附錄 2 人用前沿治療藥物的生產(chǎn)(manufacture of advanced therapy medicinal products for human use)中也明確要求對涉及傳染性病毒載體的生產(chǎn)活動需要基于書面化的污染控制策略和質(zhì)量風險管理原則采取隔離措施[3]。關于無菌藥品污染控制策略的研究對無菌藥品生產(chǎn)企業(yè)提前識別并控制污染,保證產(chǎn)品質(zhì)量有著重要的作用,對提高各類涉及無菌藥品檢查及自查工作中發(fā)現(xiàn)問題與風險的能力具有重要的意義。
1、無菌藥品污染及污染控制策略概述
對于無菌藥品常見的污染源主要包括微生物,細胞碎片(如熱原、細菌內(nèi)毒素),微粒(如玻璃、毛發(fā))[4]和病毒等,其傳播媒介一般包括氣體、水、表面、物料、人員等,其中人員占比最高(約 35%)[5]。污染控制策略(contamination control strategy ,CCS)是根據(jù)對當前產(chǎn)品及工藝在確保產(chǎn)品質(zhì)量和工藝性能方面的理解,所采取針對微生物、熱原和微粒等污染源的一系列有計劃的控制措施[6]。對無菌藥品來說污染控制策略并不是新提出的要求,現(xiàn)行藥品 GMP 中就藥品生產(chǎn)各方面的污染控制要求已經(jīng)有了明確的規(guī)定。
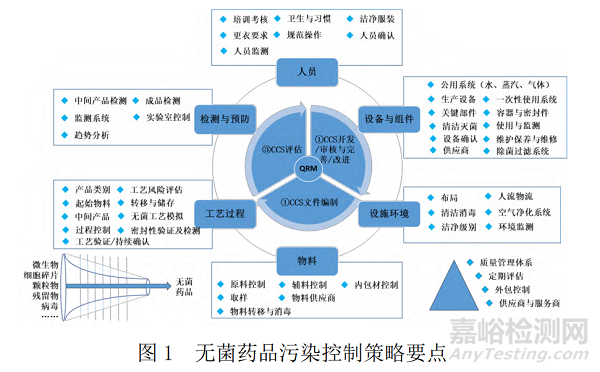
目前,EMA 新修訂的無菌藥品 GMP 附錄中對于污染控制策略提出的新要求僅是要求企業(yè)形成一份污染控制策略文件,將無菌藥品污染控制的所有關鍵控制點及控制措施有效性關聯(lián)起來進行詳細評估與闡述。這份污染控制策略文件應被納入企業(yè)質(zhì)量管理體系中,首先基于質(zhì)量風險管理(quality risk management, QRM)原則對所有污染控制要素進行系統(tǒng)全面的設計或評估,建立或進一步完善控制措施將風險消除或降低至可接受的水平(CCS 開發(fā)、審核與完善、改進);其次,在此基礎上通過對所有污染控制措施及相應文件進行關聯(lián)索引與闡述,編制污染控制策略文件(CCS 文件編制,可參考 ECA “如何開發(fā)并編制污染控制策略”中所附模板);最后,對已建立的污染控制策略進行監(jiān)測與評估(包括在管理評審中的評估),適時采取改進措施并修訂相應污染控制策略文件(CCS 評估)[6]。污染控制策略文件是一份類似于驗證主計劃、現(xiàn)場主文件的質(zhì)量體系文件,其在對無菌產(chǎn)品各要素污染控制措施進行整體分析與介紹的基礎上,涉及大量相關質(zhì)量管理體系文件的索引,以證實所有污染控制策略及其要素均已得到有效的實施。無菌藥品污染控制策略要點可參見圖 1。
2、無菌藥品污染控制策略要點分析
藥品污染控制是一個系統(tǒng)工程,在無菌藥品污染控制策略中的污染控制要素可以結合實際情況從不同的角度進行分類與闡述。本研究主要基于質(zhì)量管理理論,從影響藥品質(zhì)量的六要素進行分類,結合近幾年我國及全球其他藥品監(jiān)管檢查部門(主要包括世界衛(wèi)生組織、美國食品藥品監(jiān)督管理局和歐洲藥品監(jiān)督管理局)在無菌化學藥品及生物制品檢查中發(fā)現(xiàn)的典型問題與新情況,圍繞無菌藥品污染控制策略要點與問題實例進行分析。
2.1 人員污染控制
人員是藥品生產(chǎn)中最大的污染源。所有進入潔凈生產(chǎn)區(qū)域及生產(chǎn)相關人員必須經(jīng)過適宜的培訓并通過相應的考核(包括微生物知識及無菌行為規(guī)范的培訓),確保潔凈區(qū)內(nèi)人員的規(guī)范操作,建立進入生產(chǎn)區(qū)域人員的衛(wèi)生規(guī)范和身體不適報告制度及適宜的健康監(jiān)測要求,并對行為的規(guī)范性進行監(jiān)測,明確各潔凈房間內(nèi)最大允許進入人員數(shù)量,配備與對應潔凈環(huán)境級別和生產(chǎn)操作相匹配的潔凈工作服,制定潔凈工作服清潔、滅菌、使用次數(shù)和期限的規(guī)定并完成相應驗證。對于進入 B 級潔凈區(qū)的人員需要進行潔凈更衣培訓與更衣確認,待考核與確認合格后才能進行 B 級潔凈區(qū),建立進入 B 級潔凈區(qū)人員名單,進行人員資質(zhì)監(jiān)控與定期的再確認,相關人員按要求參與必要的無菌工藝模擬試驗,最大限度的控制人員導致的污染。
人員污染控制方面的一些典型問題包括:①潔凈區(qū)內(nèi)人員的不規(guī)范操作,如檢查期間發(fā)現(xiàn)操作人員在 B 級潔凈區(qū)內(nèi)聚集聊天、雙手抱胸、快速行走、大幅度動作、從地板上撿起物品、身體上半身或全手臂侵入 A 級區(qū)域上方操作、倚靠在生產(chǎn)設備或墻壁上、手指直接接觸控制面板、用手直接擰門把手開關門、操作后及進行關鍵操作前未進行手部消毒、跪爬進入傳遞箱傳遞物品、開關門操作中潔凈服接觸到 A 級區(qū)隔離門內(nèi)表面,以及在質(zhì)量控制潔凈區(qū)內(nèi)操作人員存在蹲坐及躺臥在地面等各類不規(guī)范行為。②不規(guī)范的潔凈服著裝,B 級區(qū)操作人員部分身體及皮膚裸露,分裝操作人員僅穿戴一層手套且手套手臂部分較短,部分潔凈服接觸到地面等。③部分管理規(guī)定缺少驗證數(shù)據(jù)支持,制度執(zhí)行存在不足。如房間最大進入人員數(shù)量、潔凈服最大清洗滅菌次數(shù)及有效期、潔凈服清洗方法等未進行對應驗證或確認;在程序執(zhí)行中存在現(xiàn)場未標識房間最大進入人數(shù),人員進入數(shù)量超過規(guī)定最大數(shù)量,潔凈服清潔滅菌記錄與滅菌器使用記錄中同時間內(nèi)滅菌套數(shù)不一致,無潔凈服滅菌記錄等問題。④相關記錄缺失或記錄信息不完整,如未建立進入 B 級區(qū)人員清單、進入 B 級區(qū)人員清單未顯示人員確認合格時間及有效期。⑤其他方面,存在未建立有效的身體不適報告制度(如缺少報告記錄等要求),進入潔凈區(qū)人員衛(wèi)生相關規(guī)定不充分(如未考慮限制患有足癬、嚴重頭皮屑等皮膚疾患者員的進入)的問題。
2.2 設備與組件污染控制
設備與組件主要包括藥品生產(chǎn)設備(包括制藥設備、制藥用水系統(tǒng)、純蒸汽系統(tǒng)、壓縮空氣系統(tǒng)、氮氣系統(tǒng)等)和與藥品直接接觸的關鍵組件及密封件(如膠塞料斗、硅膠管、物料及中間產(chǎn)品容器、密封橡膠圈等)。藥品生產(chǎn)設備需合理設計并完成確認確保持續(xù)符合預定用途,在設備使用過程中做好監(jiān)測、清潔、消毒、滅菌、維護與維修,按要求完成清潔、消毒及滅菌驗證。關鍵組件及密封件的清潔滅菌方式、貯存方法、最長貯存期限、允許使用次數(shù)(必要時)應經(jīng)過驗證。對于污染有直接影響的生產(chǎn)設備部件需要基于風險加強監(jiān)測、提高更換頻次,例如隧道烘箱中的高效過濾器、濕熱滅菌柜中的空氣濾芯、反應罐上 的 呼 吸 閥 、 隔 離 系 統(tǒng) (restricted access barrier systems, RABS)上的操作手套等。根據(jù)生產(chǎn)設備情況選擇適宜的清潔劑與消毒劑,開展消毒劑效力驗證,基于驗證結果制定科學的消毒方法。對于需除菌過濾的氣體和液體應開展相應的除菌過濾驗證,在日常生產(chǎn)工作落實過濾器完整性檢測、滅菌與使用要求。對于關鍵部件(如一次性系統(tǒng)、濾芯等)需加強供應商的管理,并建立適宜的質(zhì)量確認要求(涉及安全性、產(chǎn)品相容性等),結合一次性使用系統(tǒng)的特殊性評估其污染的風險。
設備與組件污染控制方面的一些典型問題包括:①設備設計存在潛在污染的風險,如灌裝線一側靠墻布局(<30cm)難以進行有效清潔、灌裝線 RABS 需要開門后才能進行環(huán)境監(jiān)測平皿的更換、灌裝前設備組裝需要過長時間、A 級區(qū)設備表面存在大量難以清潔的網(wǎng)孔(且企業(yè)不能證明類似孔洞有利于氣流流型的改善)、灌裝線上的膠塞料斗無法拆除進行濕熱滅菌等。②制藥用水系統(tǒng)監(jiān)測頻次不合理,如每月僅進行一次注射用水使用點的監(jiān)測、部分使用點監(jiān)測項目缺少細菌內(nèi)毒素及微生物限度。③每半年才對直接接觸物料/產(chǎn)品的壓縮氣體進行一次監(jiān)測,監(jiān)測頻率缺少有效的風險評估。④設備表面微生物監(jiān)測中,缺少針對不規(guī)則設備表面的微生物取樣方法、取樣點的選擇缺少有效的風險評估、未建立輪換/隨機表面微生物取樣位置的規(guī)定、未對 RABS 內(nèi)操作手套進行每班次的表面微生物取樣檢測。⑤關鍵性生產(chǎn)設備確認不充分,日常監(jiān)測存在不足,如:高壓滅菌器設備確認未考慮最差條件,層析介質(zhì)未開展壽命驗證,洗瓶機確認中未包括清洗針頭在瓶內(nèi)的位置規(guī)定,每月進行一次滅菌器呼吸閥完整性檢測但缺少再次使用前呼吸器組裝程序的規(guī)定,隧道烘箱確認時細菌內(nèi)毒素挑戰(zhàn)試驗點未放在安瓿瓶內(nèi)部,規(guī)定隧道烘箱每年僅進行一次高效過濾器完整性檢測且缺少合理說明,規(guī)定隧道烘箱每半年進行一次干預性監(jiān)測但再次生產(chǎn)前未對冷卻段進行消毒/熏蒸,隧道烘箱冷卻段與灌裝線之間缺少壓差監(jiān)測與記錄、內(nèi)部 3 個不同功能段之間缺少的合理壓差設計與監(jiān)控等。⑥ 在消毒劑選擇與驗證方面,存在消毒劑選擇不合理,未進行消毒劑效力驗證或驗證不充分[7](如表面材質(zhì)選擇不足、未使用環(huán)境中分離菌株、僅進行了實驗室研究等),未基于消毒劑效力驗證制定消毒操作規(guī)定,未開展層析柱消毒及保存驗證等問題。⑦清潔驗證不充分,在限度標準、清潔方法、最差條件挑戰(zhàn)性試驗、清潔驗證取樣等方面存在不足[8],如層析系統(tǒng)清潔驗證缺少對微生物限度及細菌內(nèi)毒素的評估、殘留檢測項目總有機碳標準缺少與蛋白殘留的對應關系分析、未對非專用濾芯及硅膠軟管進行清潔驗證等。⑧除菌過濾系統(tǒng)方面,存在未進行除菌過濾驗證、委托濾器供應商開展的除菌過濾驗證未能代表實際過濾工藝、對關鍵過濾操作未進行滅菌后使用前完整性檢測,過濾器完整性檢測不合格的調(diào)查處理不充分且缺少程序規(guī)定、氣體過濾器完整性檢測間隔周期較長且缺少有效評估等問題。⑨ 直接接觸物料與中間產(chǎn)品的工器具消毒滅菌不充分,如配制工序使用的量筒、不銹鋼桶、不銹鋼攪拌棒等直接接觸物料及中間產(chǎn)品的工具在 D 級潔凈區(qū)進行清洗、烘干與存放,未評估熱原及微生物的影響。⑩用于貯存中間產(chǎn)品的一次性使用塑料儲存袋在使用后未進行完整性檢測。?生產(chǎn)設備未能進行有效的維護與維修,如灌裝加塞機隔離門變形導致不能完全關閉。
2.3 設施環(huán)境污染控制
設施環(huán)境應基于生產(chǎn)藥品的特性和潔凈度級別要求進行合理的設計、布局、驗證、使用與維護,配置適宜的暖通空調(diào)系統(tǒng)(heating, ventilation and air-conditioning systems,HVAC)以保證藥品生產(chǎn)環(huán)境持續(xù)符合相應潔凈級別要求(包括屏蔽系統(tǒng)、隔離器等),盡可能的減少潔凈區(qū)內(nèi)潛在污染風險(如設施表面、夾層探頭、開關門方式、管線布局等),建立合理的潔凈區(qū)人流通道及物流通道(包括氣鎖間、互鎖要求等),保證不同潔凈度區(qū)域及有特殊控制要求區(qū)域的壓差范圍,建立有效的潔凈區(qū)內(nèi)設施環(huán)境的清潔、消毒、滅菌及監(jiān)測方式與頻次。結合最近幾年檢查發(fā)現(xiàn)問題分析,在污染控制中需要特別關注 2 個方面的內(nèi)容:一是科學合理的設施環(huán)境布局與設計,例如與 B級潔凈區(qū)域直接相連接的房間潔凈級別要求、外部對 B 級區(qū)域內(nèi)部觀察的要求、潔凈區(qū)門及開關門的設計、盡可能最靠近灌裝線位置進行除菌過濾的要求、以及潔凈區(qū)域內(nèi)氣流方式的設計等;二是基于風險的潔凈區(qū)環(huán)境監(jiān)測,環(huán)境監(jiān)測本身也是一種干預行為,故環(huán)境監(jiān)測并非越多越好,如同對驗證、質(zhì)量成本等研究一樣,關鍵點在于找到科學的平衡區(qū)間,需要基于風險原則確定監(jiān)測點數(shù)量(考慮面積、人流與物流、生產(chǎn)活動風險、布局、法規(guī)要求等),監(jiān)測項目(如塵埃粒子、浮游菌、沉降菌及表面微生物等),監(jiān)測位置(考慮房間布局、生產(chǎn)工藝、早期的網(wǎng)格設計等,常見的位置如灌裝線中瓶敞開的區(qū)域,門把手、墻及簾子的設施表面,灌裝線、控制面板、膠塞料斗、灌裝針、灌裝線操作手套等設備表面),監(jiān)測頻次(應能發(fā)現(xiàn)潛在的系統(tǒng)問題及對產(chǎn)品的潛在污染風險),并形成正式的風險評估文件,在制定過程中需要充分評估設施環(huán)境對產(chǎn)品質(zhì)量產(chǎn)生不利影響可能性、實際生產(chǎn)中的最差條件、日常監(jiān)測點的輪換及隨機監(jiān)測點的考慮、最難清潔消毒的位置、環(huán)境取樣對環(huán)境與產(chǎn)品的影響等因素,并基于歷史監(jiān)測情況確定合理的警戒限度與行動限度,進行定期回顧與評估確保當前環(huán)境監(jiān)測措施的有效性。
設施環(huán)境污染控制方面的一些典型問題包括:①在設施環(huán)境的設計方面,存在關鍵生產(chǎn)設備(如灌封線)自 B 級室內(nèi)采風但未評估對氣流流型的影響、針對部分 B 級關鍵操作區(qū)域未結合實際生產(chǎn)情況評估延展層流的必要性、潔凈房間內(nèi)送風與回風口的位置設計難以保證對房間全部區(qū)域的空氣交換、B 級潔凈區(qū)門存在難以清潔的位置且門把手設計需要操作人員用手直接進行開關操作、閉門器存在潤滑劑脫落污染環(huán)境的風險、B 級操作區(qū)域缺少外部監(jiān)測視窗或有效的監(jiān)控攝像頭、B 級關鍵操作區(qū)物料通道直接與 CNC 區(qū)域相聯(lián)通、產(chǎn)品未在盡量接近分裝點的位置進行除菌過濾且缺少合理評估及正式評估報告等問題。②在設施環(huán)境的使用與維護方面,主要存在維護和使用過程監(jiān)測不充分的問題,如現(xiàn)場發(fā)現(xiàn)潔凈區(qū)內(nèi)設施表面不光滑(破損、開膠、老化、凹陷、凸起、殘留粘合劑等),未考慮生產(chǎn)區(qū)域的關鍵性且未基于質(zhì)量風險管理原則的情況下對高效過濾器每年才進行一次完整性檢測、物流緩沖間互鎖時間設置未結合空調(diào)系統(tǒng)驗證時房間自凈時間及消毒劑最小作用時間等。③潔凈環(huán)境驗證方面,涉及氣流流型測試僅在靜態(tài)條件下開展、未能提供氣流流型視頻、氣流流型視頻顯示內(nèi)容未能證明單向流情況,驗證項目缺失(如未包括表面微生物、自凈時間等),驗證環(huán)境監(jiān)測點設置不當?shù)葐栴}。④潔凈環(huán)境監(jiān)測方面,主要是對監(jiān)測位置、監(jiān)測項目、監(jiān)測時間和監(jiān)測頻率等未結合實際生產(chǎn)及設施情況基于風險進行充分的評估與設計,存在不能及時發(fā)現(xiàn)污染情況的風險,一些實例包括:表面微生物監(jiān)測項目不包括 B 級區(qū)域,對 A 級區(qū)未包括全部關鍵位置(如灌裝機操作手套、灌裝區(qū)域和加塞區(qū)域)等,生產(chǎn)期間整個 B級區(qū)域僅對灌裝間的一個點進行浮游菌和沉降菌的監(jiān)測、A級和 B 級區(qū)浮游菌僅生產(chǎn)開始前和結束后進行監(jiān)測、C 級區(qū)域僅每月進行一次監(jiān)測、D 級區(qū)域僅每年進行一次監(jiān)測、未對潔凈環(huán)境中檢出的微生物進行菌種鑒別及采取必要的環(huán)境消毒措施。⑤環(huán)境監(jiān)測的不規(guī)范操作,如在臨近沉降菌平皿周圍上方進行消毒劑噴灑消毒、浮游菌檢測中培養(yǎng)基平皿蓋未打開、沉降菌檢測時將平皿蓋斜放在平皿上遮擋部分平皿采樣面積等。⑥ 其他廠房設施相關的污染風險,包括煙感裝置、緊急逃生門、防蟲防鼠措施、B 級區(qū)內(nèi)部大量使用會產(chǎn)生纖維的紙質(zhì)記錄等情況。⑦廠房設施消毒相關的問題與設備基本類似,不再重復描述。
2.4 物料污染控制
物料主要指藥品生產(chǎn)使用的原料、輔料和內(nèi)包裝材料。所有物料的供應商(包括生產(chǎn)商和經(jīng)銷商)應經(jīng)過確認并確保持續(xù)處于有效狀態(tài)(涉及供應商評估、供應商審計、合格供應商管理等),在整個物料供應鏈中應基于質(zhì)量風險管理的原則識別并控制關鍵環(huán)節(jié),嚴格控制物料接受、取樣、檢驗、放行、轉移及消毒等各個環(huán)節(jié)(包括必要的驗證),特別是物料的微生物負荷、細菌內(nèi)毒素及無菌要求,同時需盡量減少材料變異或受損的風險,如西林瓶及安瓿瓶等易碎內(nèi)包裝材料的運輸、轉移、清洗、干熱除熱原、灌裝、裝載、卸載、扎蓋等環(huán)節(jié)[9]。
物料污染控制方面的一些典型問題包括:①對關鍵物料運輸與轉移過程的風險控制措施不足,例如某配制用無菌物料采用單層塑料桶包裝且需海外長途運輸,未識別并有效控制運輸過程包裝破損的風險及進入潔凈區(qū)前未能充分消毒的風險;缺少對進入潔凈區(qū)物料的消毒程序驗證等。②倉庫物料收發(fā)區(qū)缺少有效設施(如足夠面積的雨棚)避免惡劣天氣對物料的影響。③取樣環(huán)節(jié)污染,存在取樣工具未進行滅菌或滅菌后缺少效期標識、取樣用潔凈服在 QC 實驗室普通實驗區(qū)域進行清洗滅菌與晾干、缺少有效措施防止粉末物料取樣時的污染(特別是活性炭)等。④物料標準中未建立微生物或細菌內(nèi)毒素檢測項目,或在缺少充分數(shù)據(jù)及合理評估的基礎上降低了相應項目的檢測頻次。⑤ 對于需進行無菌檢測的桶裝物料,未按已建立程序規(guī)定進行取樣與放行,未待無菌檢測結果提前放行使用。
2.5 生產(chǎn)工藝過程污染控制
生產(chǎn)企業(yè)應結合不同類別產(chǎn)品生產(chǎn)工藝(如非最終滅菌和最終滅菌)的差異對每個無菌產(chǎn)品生產(chǎn)工藝進行質(zhì)量風險評估,開展工藝驗證(包括持續(xù)工藝確認),同時對用于滅菌、消毒、病毒去除或滅活的方法進行驗證,并采取有效措施避免再次污染的風險。在生產(chǎn)全過程嚴格起始物料、中間產(chǎn)品、過程轉移與儲存、過程控制、各工序的無菌操作及干預(包括包裝環(huán)節(jié)[10])、對密封性驗證及檢測[11]、目檢等方面可能的污染及污染控制措施進行管控,對非最終滅菌產(chǎn)品的無菌生產(chǎn)工藝全過程進行無菌工藝模擬,確保生產(chǎn)工藝過程中的污染得到了有效控制。生產(chǎn)過程中,除微生物、細胞碎片和病毒等污染外,還需特別關注顆粒物(包括玻璃、橡膠、金屬、普通纖維、塑料纖維和毛發(fā)等)的污染控制,特別是全自動設備的應用、驗證和日常監(jiān)測[12]。以凍干粉針劑生產(chǎn)工藝過程中的污染控制為例,可以從生產(chǎn)前清洗和準備、藥液配制、除菌過濾、灌裝、半壓塞及自動進箱、冷凍干燥、出箱軋蓋、目檢、貼簽和包裝等各工序的污染控制措施進行分析與控制[13]。生產(chǎn)工藝過程污染控制方面的一些典型問題包括:①中間產(chǎn)品控制中,存在配制后的中間產(chǎn)品除菌過濾前未進行微生物負荷檢測及檢測標準制定不合理的情況。②過程轉移與儲存中,存在未進行中間產(chǎn)品存放時間驗證或驗證時間不足、中間產(chǎn)品經(jīng) CNC 區(qū)域進入 B 級區(qū)的表面清潔消毒程序未經(jīng)驗證、無菌生產(chǎn)現(xiàn)場裝有中間產(chǎn)品的玻璃瓶缺少狀態(tài)標識等問題。③無菌工藝模擬中,主要問題是未能有效模擬實際生產(chǎn)工藝及無菌操作全過程,同時也存在模擬試驗記錄內(nèi)容不完整、干預操作設計不合理、未按規(guī)定每班次每半年至少進行一次無菌工藝模擬、一年未生產(chǎn)的非常規(guī)生產(chǎn)首次啟動時未進行連續(xù) 3 次成功的無菌工藝模擬等問題。④密封性驗證及檢測中,包括工作細胞冷凍保存用的帶螺帽冷凍管未進行密封性驗證或檢測,除菌過濾后原液存放的帶螺帽玻璃瓶容器未進行密封性驗證,最終成品未進行密封性驗證或驗證不充分等。⑤燈檢工序中,存在燈檢機照度不符合標準規(guī)定、燈檢機照度監(jiān)測位置未包括實際燈檢區(qū)域、燈檢人員燈檢期間休息時間規(guī)定難以保證眼部疲勞的恢復等。⑥ 去除/滅活病毒能力驗證中,如 37%甲醛滅活病驗證方案缺少攪拌速率、溫度信息,滅活前未進行病毒聚集試驗,滅活病毒驗證的裝載模式與日常生產(chǎn)中滅活病毒的裝載模式不一致等。
2.6 檢測在污染控制中的要求
盡管無菌檢測的檢出能力由于檢測數(shù)量的統(tǒng)計學因素導致檢出能力較低,以一個直觀的計算為例:生產(chǎn)了 100000 支注射劑,其中有 1000 支染菌(不合格率 1%),隨機抽取 40 支進行規(guī)范的無菌檢測,無菌檢測合格的概率約為 66.9%。取樣檢測的局限性在無菌檢測有較為明顯的體現(xiàn)。盡管如此,無菌檢測仍起到重要的把關作用,特別當批污染率大于 15%時,無菌檢測不合格概率已大于 99.8%。進一步結合科學的取樣策略,能更加有效的提高無菌檢測在污染識別中的效力。整體上看,檢測對污染的控制起著重要的作用,主要包括在良好實驗室控制下的中間產(chǎn)品檢測、成品檢測、趨勢分析(如制藥用水系統(tǒng)數(shù)據(jù)趨勢、潔凈區(qū)環(huán)境監(jiān)測趨勢、中間產(chǎn)品及成品檢測趨勢等),通過對生產(chǎn)環(huán)境(潔凈區(qū)與 CNC 區(qū)域)、原輔料、生產(chǎn)人員、制藥用水中監(jiān)測到微生物的鑒別與數(shù)據(jù)的累積,為藥品生產(chǎn)過程污染微生物的溯源分析調(diào)查提供數(shù)據(jù)依據(jù)[14],是藥品微生物過程控制中的重要內(nèi)容[15]。
從Pre-IND到NDA上市申請的全程法規(guī)與實操及案例分析
污染控制中在檢測方面的一些典型問題包括:①無菌及微生物檢測用培養(yǎng)基控制不足,如檢查發(fā)現(xiàn)現(xiàn)場培養(yǎng)中的成品檢測硫乙醇酸鹽流體培養(yǎng)基樣品瓶(集菌器內(nèi))已全部氧化,微生物限度檢測用培養(yǎng)基未進行促生長試驗等。②超標及超趨勢結果調(diào)查處理不充分, 如企業(yè)未能對報告和記錄虛假環(huán)境監(jiān)測結果根本原因進行適宜的調(diào)查等。③對環(huán)境監(jiān)測或制藥用水的趨勢分析中未能識別并有效控制異常趨勢或情況,如環(huán)境監(jiān)測發(fā)現(xiàn)在 B 級灌裝間操作人員潔凈服額頭位置表面檢出表面微生物(1cfu/皿),企業(yè)未評估在潔凈服表面的微生物暴露在層流下時可能最終污染關鍵表面(如敞口西林瓶內(nèi))的風險,并對相關批次進行了放行;未對注射用水微生物限度檢測超過警戒限的情況進行分析評估。④環(huán)境監(jiān)測平皿的觀察結果缺少其他人員對平皿的復核。
3、結束語
污染控制策略的核心是基于可能的污染來源,針對藥品生產(chǎn)質(zhì)量管理相關要素建立一系列的控制措施及過程控制方案,明確所有的關鍵控制點和控制措施,避免產(chǎn)品的污染。無菌藥品的污染控制不能僅依靠某一項控制措施,其取決于系統(tǒng)性的污染控制策略。本文在對無菌藥品污染控制策略生命周期管理及文件要求進行分析的基礎上,對主要污染源及污染控制措施方面的典型缺陷實例進行了總結提煉,可為無菌藥品生產(chǎn)企業(yè)進一步加強藥品污染控制提供參考,不斷降低藥品污染風險,持續(xù)保證藥品質(zhì)量。同時,這些典型問題也是近年來國內(nèi)外無菌藥品生產(chǎn)質(zhì)量管理的污染控制中相對集中且容易被忽視的內(nèi)容,也可以為無菌藥品檢查工作提供借鑒。