您當(dāng)前的位置:檢測(cè)資訊 > 科研開(kāi)發(fā)
嘉峪檢測(cè)網(wǎng) 2025-04-13 14:00
摘 要
隨著我國(guó)藥品審評(píng)審批制度的不斷完善和臨床試驗(yàn)國(guó)際化進(jìn)程的加快,I 期臨床試驗(yàn)中健康參與者的安全性管理成為一項(xiàng)關(guān)鍵挑戰(zhàn)。為應(yīng)對(duì)這一需求,本共識(shí)基于國(guó)內(nèi)多家資深臨床試驗(yàn)機(jī)構(gòu)及申辦方的實(shí)踐經(jīng)驗(yàn),結(jié)合《藥物臨床試驗(yàn)不良事件相關(guān)性評(píng)價(jià)技術(shù)指導(dǎo)原則(試行)》、國(guó)內(nèi)外相關(guān)法規(guī)及標(biāo)準(zhǔn)等,提出了一套系統(tǒng)性的健康參與者安全性管理策略。本共識(shí)重點(diǎn)涵蓋健康參與者的定義及篩選入組標(biāo)準(zhǔn)、給藥后安全性評(píng)價(jià),以及方案設(shè)計(jì)和實(shí)施中的安全性風(fēng)險(xiǎn)管控措施等內(nèi)容。通過(guò)總結(jié)實(shí)踐經(jīng)驗(yàn)與優(yōu)化管理方法,本共識(shí)旨在規(guī)范健康參與者的安全管理流程,提升I 期臨床試驗(yàn)的安全性和科學(xué)性,為國(guó)內(nèi)外同行提供參考。
With the continuous improvement of China's drug review and approval system and the acceleration of clinical trial internationalization, the safety management of healthy participants in phase I clinical trials has become a critical challenge. In response, this consensus draws on the practical experience of multiple leading domestic clinical trial institutions and sponsors, incorporating the Technical Guidelines for the Causality Assessment of Adverse Events in Drug Clinical Trials (Provisional) and relevant domestic and international regulations and standards, to propose a systematic safety management strategy for healthy participants. This consensus focuses on the definition and screening criteria for healthy participants, post-dosing safety evaluation, and safety risk management measures in protocol design. By summarizing practical experiences and optimizing management approaches, this consensus aims to standardize the safety management processes for healthy participants,enhance the safety and scientific rigor of phase I clinical trials, and provide a reference for both domestic and international peers.
關(guān)鍵詞
I 期臨床試驗(yàn);健康參與者;安全性管理;風(fēng)險(xiǎn)管控;策略
phase I clinical trials; healthy participants; safety management; risk control; strategy
近年來(lái),隨著我國(guó)藥品審評(píng)審批制度改革以及加入國(guó)際人用藥品注冊(cè)技術(shù)協(xié)調(diào)會(huì)(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH) 后, 藥品審評(píng)審批制度持續(xù)深化和改革創(chuàng)新,監(jiān)管體系不斷完善并逐漸與國(guó)際接軌,醫(yī)藥創(chuàng)新政策環(huán)境得到明顯改善。隨之,全球首次人體Ⅰ期臨床試驗(yàn)的需求在我國(guó)越來(lái)越多。Ⅰ期臨床試驗(yàn)是初步的臨床藥理學(xué)及人體安全性評(píng)價(jià)試驗(yàn),通過(guò)觀察人體對(duì)于新藥的耐受程度和藥代動(dòng)力學(xué)(pharmacokinetic,PK)特征,為制定給藥方案提供依據(jù)。多數(shù)Ⅰ期臨床試驗(yàn)以健康參與者為研究對(duì)象,而其安全性管理存在巨大挑戰(zhàn)。
最新發(fā)布實(shí)施的ICH《E6(R3):藥物臨床試驗(yàn)質(zhì)量管理規(guī)范》[E6(R3): Guideline for Good Clinical Practice] 中,強(qiáng)調(diào)了基于風(fēng)險(xiǎn)的臨床試驗(yàn)質(zhì)量管理理念。2024 年6 月國(guó)家藥監(jiān)局藥品審評(píng)中心(以下簡(jiǎn)稱(chēng)藥審中心)發(fā)布了《藥物臨床試驗(yàn)不良事件相關(guān)性評(píng)價(jià)技術(shù)指導(dǎo)原則(試行)》,規(guī)范我國(guó)藥物臨床試驗(yàn)中不良事件與試驗(yàn)藥物相關(guān)性評(píng)價(jià)方法與標(biāo)準(zhǔn),以更好地實(shí)現(xiàn)臨床試驗(yàn)風(fēng)險(xiǎn)最小化,保護(hù)參與者安全。目前國(guó)內(nèi)外尚缺乏統(tǒng)一的健康參與者安全性管理標(biāo)準(zhǔn)或指南。基于此, 我國(guó)4 家數(shù)年來(lái)從事多項(xiàng)創(chuàng)新藥I 期臨床試驗(yàn)的資深臨床試驗(yàn)機(jī)構(gòu)研究者,聯(lián)合中國(guó)外商投資企業(yè)協(xié)會(huì)藥品研制和開(kāi)發(fā)工作委員會(huì)(China Associationof Enterprises with Foreign Investment R&D-based Pharmaceutical Association Committee,RDPAC) 部分會(huì)員企業(yè)專(zhuān)業(yè)人士代表,結(jié)合自身臨床試驗(yàn)實(shí)踐經(jīng)驗(yàn)并參照國(guó)內(nèi)外相關(guān)法規(guī)及標(biāo)準(zhǔn)等,形成本共識(shí),供同行參考。
1.目的
制定健康參與者Ⅰ期臨床試驗(yàn)中的安全性風(fēng)險(xiǎn)管控要點(diǎn),保障參與者安全;明確Ⅰ期臨床試驗(yàn)中健康參與者給藥后安全性評(píng)價(jià)的關(guān)鍵要素;推薦健康參與者篩選入組標(biāo)準(zhǔn)的考量要點(diǎn),降低不同項(xiàng)目或研究機(jī)構(gòu)安全性評(píng)估差異性所導(dǎo)致的參與者安全性風(fēng)險(xiǎn)。
2. 適用范圍
包括但不限于首次人體試驗(yàn)、生物等效性、食物影響、藥物間相互作用研究等以健康參與者為對(duì)象的臨床藥理學(xué)研究。
3. 主要內(nèi)容
3.1 健康參與者概念
目前, 健康參與者尚無(wú)明確定義,世界衛(wèi)生組織(World Health Organization,WHO)將健康定義為完全的身體、精神和社會(huì)的狀態(tài),而不僅僅是沒(méi)有疾病或殘疾[1]。臨床試驗(yàn)中的健康參與者通常是指那些沒(méi)有明顯疾病或健康問(wèn)題的人群,同時(shí)其精神狀態(tài)能夠保證正確理解并自愿參與研究、簽署知情同意書(shū)[2-3]。部分研究可能會(huì)納入個(gè)別指標(biāo)異常的參與者,但這種異常不代表健康狀況異常,且和參與試驗(yàn)的目的、可能導(dǎo)致的風(fēng)險(xiǎn)等無(wú)關(guān)。每項(xiàng)試驗(yàn)會(huì)根據(jù)試驗(yàn)?zāi)康脑谠囼?yàn)方案中確定健康參與者的入選和排除標(biāo)準(zhǔn)。
3.2 健康參與者篩選入組標(biāo)準(zhǔn)
根據(jù)2021 年12 月國(guó)家藥監(jiān)局食品藥品審核查驗(yàn)中心發(fā)布的《藥品注冊(cè)核查要點(diǎn)與判定原則(藥物臨床試驗(yàn))(試行)》,參與者在篩選入組、給藥、隨訪(fǎng)全過(guò)程中的安全性評(píng)估、醫(yī)學(xué)判斷和臨床決策,必須由機(jī)構(gòu)具有執(zhí)業(yè)資格的醫(yī)學(xué)專(zhuān)業(yè)人員執(zhí)行,確保專(zhuān)業(yè)性和合法性,不可由非醫(yī)學(xué)背景的研究者或協(xié)調(diào)員或申辦者擅自進(jìn)行醫(yī)學(xué)判斷或干擾研究醫(yī)生的醫(yī)學(xué)判斷。健康參與者的臨床試驗(yàn)要求將風(fēng)險(xiǎn)最小化,因此需要根據(jù)試驗(yàn)藥物臨床前、已完成臨床試驗(yàn)(如有)或同靶點(diǎn)藥物安全性信息等,對(duì)于毒性靶器官相關(guān)的指標(biāo),制定嚴(yán)格的篩選入組標(biāo)準(zhǔn)。雖然每個(gè)方案都有明確的入選及排除標(biāo)準(zhǔn),但很難囊括所有指標(biāo)細(xì)則,特別是實(shí)驗(yàn)室指標(biāo)異常達(dá)到何種程度時(shí)不能入組,通常由每家研究中心考量,國(guó)內(nèi)外對(duì)此尚無(wú)統(tǒng)一標(biāo)準(zhǔn)。本共識(shí)在遵從研究方案入選和排除標(biāo)準(zhǔn)、基于試驗(yàn)藥物作用機(jī)制和已有安全性信息、確保操作可行性等原則下,對(duì)部分檢查指標(biāo)給出了具體的入組建議,旨在提高篩選入組標(biāo)準(zhǔn)的統(tǒng)一性。在實(shí)際操作層面,需要研究者根據(jù)試驗(yàn)藥物的相關(guān)風(fēng)險(xiǎn)進(jìn)行整體把控,必要時(shí)可進(jìn)行調(diào)整,最根本的原則是充分保證參與者的安全。對(duì)于健康參與者篩選入組標(biāo)準(zhǔn)(主要針對(duì)客觀指標(biāo),如生命體征、心電圖參數(shù)及實(shí)驗(yàn)室檢查指標(biāo)等)的主要考量要點(diǎn),經(jīng)專(zhuān)家討論達(dá)成了一致推薦,歸納如下。
3.2.1 體格檢查
臨床試驗(yàn)中的體格檢查項(xiàng)目通常包括一般情況、頭頸部、胸部、腹部、背部、四肢、神經(jīng)系統(tǒng)、皮膚黏膜和淋巴結(jié)等檢查。根據(jù)藥物特性,部分需要專(zhuān)科查體,如神經(jīng)系統(tǒng)、眼科、耳鼻喉科、泌尿生殖系統(tǒng)等檢查。
本共識(shí)建議:①對(duì)于一些慢性病的穩(wěn)定體征,如處于穩(wěn)定期的足癬或手癬,未見(jiàn)明顯脫屑、糜爛、滲出等體征,且試驗(yàn)藥物無(wú)相關(guān)風(fēng)險(xiǎn)時(shí),可酌情放寬入組條件。而發(fā)現(xiàn)急性疾病相關(guān)體征,如局部皮膚紅腫破潰等,不建議入組。②根據(jù)試驗(yàn)藥物的藥理特性、臨床前和已完成臨床試驗(yàn)(如有)安全性風(fēng)險(xiǎn)或同靶點(diǎn)藥物安全性信息,對(duì)參與者的體格檢查進(jìn)行把控。例如,具有免疫抑制作用的大分子藥物有引起感染的風(fēng)險(xiǎn),需加強(qiáng)感染性疾病相關(guān)體征的把控,包括足癬或手癬、毛囊炎、帶狀皰疹等;靜脈用藥物,特別是已知容易引起靜脈炎的試驗(yàn)藥物,需加強(qiáng)血管評(píng)估;皮膚外用試驗(yàn)藥物,應(yīng)加強(qiáng)計(jì)劃給藥部位的評(píng)估,有皮損、脫屑、文身和疤痕等體征的參與者不能入組,以免影響給藥部位評(píng)估或增加參與者安全性風(fēng)險(xiǎn);已知有神經(jīng)系統(tǒng)安全性風(fēng)險(xiǎn)的試驗(yàn)藥物,需加強(qiáng)神經(jīng)系統(tǒng)專(zhuān)科查體,包括顱神經(jīng)、運(yùn)動(dòng)功能、感覺(jué)功能、神經(jīng)反射(包括淺反射、深反射、病理反射)等檢查。
3.2.2 生命體征及心電圖
有關(guān)健康參與者生命體征及心電圖篩選入組標(biāo)準(zhǔn)的考量要點(diǎn),見(jiàn)表1[4-20]。

3.2.3 實(shí)驗(yàn)室檢查
不伴有異常癥狀和體征的實(shí)驗(yàn)室檢查結(jié)果異常是參與者篩選失敗的最主要原因, 約占34%~47%[21-22]。其中血生化指標(biāo)異常的發(fā)生概率最高,其次是尿常規(guī)和血常規(guī)異常。血生化指標(biāo)異常主要表現(xiàn)為肝功能、血脂和血尿酸的異常。實(shí)驗(yàn)室檢查指標(biāo)若出現(xiàn)異常,達(dá)到何種水平時(shí)不能入組,一直是困擾研究者、申辦者、檢查人員的主要問(wèn)題和分歧點(diǎn)。有些實(shí)驗(yàn)室檢查指標(biāo)會(huì)受多種因素影響(如運(yùn)動(dòng)、飲食、睡眠、休息等)而出現(xiàn)生理性波動(dòng)。關(guān)于一些常見(jiàn)實(shí)驗(yàn)室檢查指標(biāo)的篩選入組標(biāo)準(zhǔn)(此處特指不伴隨癥狀或體征) 的推薦意見(jiàn), 見(jiàn)表2[8,23-44]。除了表2 已列舉的實(shí)驗(yàn)室檢查指標(biāo),尚有許多其他指標(biāo)未在此詳細(xì)說(shuō)明,這些未列出的指標(biāo),可基于試驗(yàn)藥物、研究方案及安全性風(fēng)險(xiǎn)總體把控等進(jìn)行全面評(píng)估,一般而言,其接受范圍可適當(dāng)放寬至正常參考值上限或下限的約20% 以?xún)?nèi)。

3.2.4 影像學(xué)檢查及其他特殊檢查
在胸部正位片、肺部CT、消化系統(tǒng)B 超、泌尿系統(tǒng)B 超等影像學(xué)檢查篩選階段,可能發(fā)現(xiàn)一些良性、非活動(dòng)性病變(如輕度脂肪肝、肝內(nèi)鈣化灶、膽囊息肉、肝腎囊腫、甲狀腺Ⅰ級(jí)或Ⅱ級(jí)結(jié)節(jié)等),這些病變并不影響器官功能,考慮以上情況并不增加參與者參加試驗(yàn)的風(fēng)險(xiǎn),因此本共識(shí)認(rèn)為可納入試驗(yàn)。對(duì)于甲狀腺Ⅲ級(jí)及以上結(jié)節(jié)、腎結(jié)石、膽囊結(jié)石等其他檢查異常是否可納入,建議由研究者根據(jù)試驗(yàn)藥物特性和安全性風(fēng)險(xiǎn)綜合把控,若有活動(dòng)性發(fā)作的風(fēng)險(xiǎn),則不建議入組。肺結(jié)節(jié)是肺部影像學(xué)檢查中常見(jiàn)的異常征象,其篩選入組的具體推薦意見(jiàn),見(jiàn)表1[18-20]。
3.3 參與者給藥后安全性評(píng)價(jià)
新藥的安全性評(píng)價(jià)必須嚴(yán)格遵循預(yù)定的試驗(yàn)方案執(zhí)行,而該方案的設(shè)計(jì)需根據(jù)試驗(yàn)藥物臨床前、已完成臨床試驗(yàn)(如有)或同靶點(diǎn)藥物安全性信息,預(yù)先設(shè)定安全性評(píng)估的具體內(nèi)容和時(shí)間安排。評(píng)估內(nèi)容主要包括參與者用藥前后以及不同隨訪(fǎng)時(shí)間點(diǎn)所收集的不良事件(adverse event,AE)、體格檢查記錄、生命體征監(jiān)測(cè)、實(shí)驗(yàn)室檢查結(jié)果以及PK 數(shù)據(jù)等。同時(shí),還應(yīng)根據(jù)試驗(yàn)藥物安全性特征以及試驗(yàn)執(zhí)行程序中可能潛在的風(fēng)險(xiǎn)因素,實(shí)施風(fēng)險(xiǎn)管理措施,從而全方位保障參與者的安全。在試驗(yàn)持續(xù)進(jìn)行過(guò)程中,對(duì)于新收集到的臨床數(shù)據(jù),可考慮動(dòng)態(tài)更新試驗(yàn)方案中相關(guān)安全性評(píng)估內(nèi)容[45-46]。
安全性評(píng)估內(nèi)容中,AE 是安全性數(shù)據(jù)收集和評(píng)估的重要依據(jù),下文針對(duì)AE 的定義、分級(jí)標(biāo)準(zhǔn)、與試驗(yàn)藥物的因果關(guān)系評(píng)估等方面具體展開(kāi)闡述。
3.3.1 不良事件(AE)的定義
2020 年4 月發(fā)布的《藥物臨床試驗(yàn)質(zhì)量管理規(guī)范》和2023年12 月發(fā)布的《新藥臨床安全性評(píng)價(jià)技術(shù)指導(dǎo)原則》均已對(duì)“不良事件”和“嚴(yán)重不良事件”進(jìn)行了定義,本文不再贅述。
參與者在使用試驗(yàn)藥物后,一旦出現(xiàn)任何有臨床意義的異常,應(yīng)及時(shí)記錄該異常為AE,并判斷AE 嚴(yán)重程度分級(jí)和與試驗(yàn)藥物的相關(guān)性,持續(xù)監(jiān)測(cè)AE 動(dòng)態(tài)變化直至結(jié)局。對(duì)于實(shí)驗(yàn)室檢查和輔助檢查指標(biāo)異常,研究者需要結(jié)合臨床癥狀、體格檢查和(或)對(duì)指標(biāo)的動(dòng)態(tài)監(jiān)測(cè)結(jié)果等內(nèi)容綜合判定。該部分目前尚無(wú)統(tǒng)一的標(biāo)準(zhǔn)和界限,可參考本共識(shí)3.2項(xiàng)下相關(guān)內(nèi)容進(jìn)行判讀,同時(shí)還需要結(jié)合以下4 個(gè)方面進(jìn)行綜合評(píng)估:①試驗(yàn)藥物或同靶點(diǎn)藥物已知的風(fēng)險(xiǎn)提示。②參與者給藥后指標(biāo)相較于給藥前基線(xiàn)的變化程度和波動(dòng)情況。③單次及多次給藥后指標(biāo)的動(dòng)態(tài)變化趨勢(shì),是否與劑量遞增和給藥頻率的增加有相關(guān)性。④觀察是單個(gè)受試者出現(xiàn)的指標(biāo)異常,還是多個(gè)受試者出現(xiàn)相同的指標(biāo)異常。若為個(gè)別受試者出現(xiàn)的一過(guò)性且無(wú)需臨床干預(yù)的無(wú)伴隨癥狀及體征的輕度指標(biāo)異常,可考慮判定為無(wú)臨床意義;若該異常指標(biāo)在參與者中發(fā)生率較高,需謹(jǐn)慎判斷其有無(wú)臨床意義。
3.3.2 AE 的分級(jí)標(biāo)準(zhǔn)
目前我國(guó)常用的分級(jí)標(biāo)準(zhǔn)主要包括三分量表分級(jí)標(biāo)準(zhǔn)、常見(jiàn)不良事件評(píng)價(jià)標(biāo)準(zhǔn)(common terminologycrit eria for adverse events,CTCAE) 以及預(yù)防用疫苗臨床試驗(yàn)不良事件分級(jí)標(biāo)準(zhǔn)3 種。這些分級(jí)標(biāo)準(zhǔn)可幫助研究者評(píng)估和報(bào)告試驗(yàn)中發(fā)生的不良事件,且能提高不同研究機(jī)構(gòu)間評(píng)估結(jié)果的一致性。
(1)三分量表分級(jí)標(biāo)準(zhǔn)。該分級(jí)方式將AE 分為輕度、中度和重度。其中,輕度是指較輕或一過(guò)性的不適,不需要治療或需要很少治療,且不影響參與者的日常活動(dòng)。中度是指癥狀或體征的不適可對(duì)日常活動(dòng)造成一些妨礙,需要采取治療措施。重度是指不能進(jìn)行正常的日常活動(dòng),需要采取全身性的藥物治療或其他治療措施。上述分級(jí)的相關(guān)描述可能因研究試驗(yàn)方案的不同而略有差異,該分級(jí)標(biāo)準(zhǔn)因易于理解及實(shí)施,應(yīng)用較為廣泛。該分級(jí)標(biāo)準(zhǔn)要求考慮試驗(yàn)藥物對(duì)參與者日常活動(dòng)的影響程度及影響時(shí)長(zhǎng)。一般認(rèn)為,僅在局部使用非激素藥物或進(jìn)行非藥物處理的情形,如口腔潰瘍使用鹽水漱口、低血糖癥狀發(fā)生時(shí)口服糖塊或糖水、發(fā)生皮炎時(shí)使用爐甘石洗劑等,屬于輕度AE 定義中的很少治療范圍。對(duì)于那些需要短期口服或皮膚外用激素類(lèi)藥物及靜脈合并用藥的情況,通常符合中度AE的定義。
(2) 常見(jiàn)不良事件評(píng)價(jià)標(biāo)準(zhǔn)[47]。CTCAE 是指由美國(guó)國(guó)家癌癥研究所(National Cancer Institute,NCI) 和美國(guó)國(guó)立衛(wèi)生研究院(National Institutesof Health,NIH)頒布的腫瘤藥物臨床試驗(yàn)的常見(jiàn)不良事件術(shù)語(yǔ)評(píng)價(jià)標(biāo)準(zhǔn),將 AE 嚴(yán)重程度分為1~5 級(jí),不同等級(jí)對(duì)應(yīng)不同的嚴(yán)重程度和臨床表現(xiàn)。CTCAE 涵蓋了包括生理、生化、實(shí)驗(yàn)室檢查指標(biāo)和臨床癥狀在內(nèi)的各種類(lèi)型不良事件,除在腫瘤藥物臨床試驗(yàn)中普遍應(yīng)用外, 也在其他各類(lèi)藥物臨床試驗(yàn)中廣泛應(yīng)用。AE 嚴(yán)重程度分級(jí)詳見(jiàn)CTCAE5.0 版。
(3)預(yù)防用疫苗臨床試驗(yàn)不良事件分級(jí)標(biāo)準(zhǔn)[48]。該標(biāo)準(zhǔn)分別從臨床觀察指標(biāo)(即癥狀和體征,包括接種部位、生命體征、非接種部位)及實(shí)驗(yàn)室檢查指標(biāo)(血液生化、血常規(guī)、尿常規(guī)等)兩大部分進(jìn)行分級(jí),通常用于疫苗臨床試驗(yàn)的健康成年和青少年志愿者,詳細(xì)標(biāo)準(zhǔn)可參考國(guó)家藥監(jiān)局發(fā)布的《預(yù)防用疫苗臨床試驗(yàn)不良事件分級(jí)標(biāo)準(zhǔn)指導(dǎo)原則》。
AE 嚴(yán)重程度的分級(jí)標(biāo)準(zhǔn)應(yīng)嚴(yán)格依據(jù)試驗(yàn)方案執(zhí)行,本共識(shí)推薦同一試驗(yàn)藥物的不同階段臨床研究采取同一分級(jí)標(biāo)準(zhǔn),以提高不同研究機(jī)構(gòu)之間對(duì)試驗(yàn)藥物毒性評(píng)估的一致性。同時(shí)推薦在以健康參與者為研究對(duì)象的Ⅰ期臨床試驗(yàn)中采用三分量表分級(jí)標(biāo)準(zhǔn)對(duì)AE 進(jìn)行分級(jí),即以是否需要采取治療措施及是否對(duì)參與者的正常生活造成干擾為變量進(jìn)行評(píng)估,可簡(jiǎn)化評(píng)估流程,減少不同研究機(jī)構(gòu)與研究人員間的評(píng)估差異,從而更快、更有效地識(shí)別并應(yīng)對(duì)試驗(yàn)藥物潛在的安全風(fēng)險(xiǎn)。
3.3.3 AE 與試驗(yàn)藥物的因果關(guān)系評(píng)估
3.3.3.1 一般考量及總體原則
AE 和試驗(yàn)藥物的因果關(guān)系評(píng)估是Ⅰ期臨床試驗(yàn)健康參與者安全性管理的關(guān)鍵點(diǎn)和難點(diǎn)。需要考慮多方面因素,包括參與者本身的個(gè)體因素和合并疾病、試驗(yàn)藥物的AE 數(shù)據(jù)、參與者合并用藥信息、試驗(yàn)藥物的臨床前毒理及藥理學(xué)證據(jù)、試驗(yàn)藥物的作用機(jī)制、試驗(yàn)藥物用藥時(shí)間和AE發(fā)生時(shí)間是否存在一定的時(shí)間關(guān)系、暫停和(或)恢復(fù)試驗(yàn)用藥后AE 的變化、同靶點(diǎn)藥物的安全性數(shù)據(jù)、試驗(yàn)藥物和AE 之間是否存在劑量- 暴露- 效應(yīng)關(guān)系等。
2024 年6 月藥審中心發(fā)布的《藥物臨床試驗(yàn)不良事件相關(guān)性評(píng)價(jià)技術(shù)指導(dǎo)原則( 試行)》[49]中強(qiáng)調(diào),對(duì)于個(gè)例AE 與試驗(yàn)藥物的相關(guān)性評(píng)價(jià),一般應(yīng)遵守時(shí)序性、合理性、劑量- 暴露- 效應(yīng)關(guān)系、實(shí)驗(yàn)/ 試驗(yàn)證據(jù)支持、可重復(fù)性、類(lèi)比、一致性及特異性共8 個(gè)基本原則。評(píng)價(jià)要點(diǎn)包括:試驗(yàn)用藥與AE 的出現(xiàn)是否有合理的時(shí)間關(guān)系,AE 是否符合該藥物已知的作用機(jī)制、特性或已知的不良反應(yīng),去激發(fā)和再激發(fā)的結(jié)果(如適用),以及AE 是否可以用參與者的疾病進(jìn)展(包括伴隨疾病)、合并用藥的作用、其他治療措施或干擾因素等的影響來(lái)解釋。目前,臨床試驗(yàn)中應(yīng)用較多的評(píng)價(jià)方法和標(biāo)準(zhǔn)主要包括五分法和二分法,具體內(nèi)容詳見(jiàn)該指導(dǎo)原則[49]。
各種評(píng)價(jià)方法各有優(yōu)缺點(diǎn),對(duì)于I 期臨床試驗(yàn)而言,可根據(jù)具體方案選擇合適的評(píng)價(jià)方案實(shí)施和執(zhí)行,本共識(shí)不做具體推薦。
3.3.3.2 藥物相互作用臨床試驗(yàn)中的因果關(guān)系評(píng)估
藥物相互作用(drug-drug interaction,DDI) 臨床試驗(yàn)是為了確認(rèn)體內(nèi)是否會(huì)發(fā)生DDI 及其嚴(yán)重程度[50]。由于DDI 臨床試驗(yàn)包含至少2 種藥物,在AE 因果關(guān)系判定時(shí),除了需要考慮藥物暴露時(shí)間、PK 特征、試驗(yàn)藥物藥理作用、前期非臨床及臨床研究結(jié)果等因素外[49],還具有以下特殊性:① DDI 臨床試驗(yàn)通常采用隨機(jī)交叉試驗(yàn)設(shè)計(jì),一般分為單用藥物和聯(lián)合用藥,共3 個(gè)階段(即藥物A →藥物B →藥物A+B),中間常設(shè)置清洗期。在藥物A 和藥物B 順序給藥后出現(xiàn)的AE,是否能完全排除與藥物A 的關(guān)系,除了要考慮研究藥物消除半衰期特征外,還需要特別關(guān)注藥物本身的藥理特性,尤其當(dāng)研究設(shè)計(jì)中沒(méi)有明顯的清洗期時(shí)。②在DDI 臨床試驗(yàn)中,經(jīng)常遇到在使用藥物A 或藥物B 后出現(xiàn)AE 的情況,在后續(xù)給藥或聯(lián)合用藥階段,AE 嚴(yán)重程度加重,此時(shí)因果關(guān)系判定除考慮AE 與用藥先后順序的時(shí)間關(guān)系外,還需考慮藥物合用對(duì)PK 特征改變的作用等因素。例如,對(duì)于在藥物B 多次給藥階段未發(fā)生、但在藥物B 與藥物A 聯(lián)合使用后發(fā)生的AE,若該AE 同時(shí)符合藥物A、B 的藥理特性,則在因果關(guān)系判定中,需考慮用藥后是否增加藥物A 或藥物B 的暴露量,是否有藥物蓄積等問(wèn)題,除非有充分的證據(jù),否則不能輕易排除兩種藥物與AE 的關(guān)系。
3.3.4 不同類(lèi)別藥物的一般安全性特征和AE 特殊考慮
在Ⅰ期臨床試驗(yàn),尤其是首次人體試驗(yàn)中, 了解藥物的藥理學(xué)和非臨床安全性特征對(duì)確保健康參與者的用藥安全至關(guān)重要。目前國(guó)內(nèi)外已發(fā)布了一些文件以支持臨床前的安全性評(píng)估, 主要包括:① ICH《M3(R2):支持藥物進(jìn)行臨床試驗(yàn)和上市的非臨床安全性研究指導(dǎo)原則》[M3(R2): Guidance on Nonclinical Safety Studies for the Conduct of Human Clinical Trialsand Marketing Authorization for Pharmaceuticals] 提供了針對(duì)人體臨床試驗(yàn)和藥品上市許可所需的非臨床安全性研究指導(dǎo)。② ICH《S6(R1):生物制品的臨床前安全性評(píng)價(jià)》[S6(R1):Preclinical Safety Evaluation of Biotechnology - Derived Pharmaceuticals] 提供了有關(guān)生物技術(shù)衍生藥物(生物制劑)特定要求的其他信息。③ ICH《S9:抗腫瘤藥物非臨床評(píng)價(jià)指導(dǎo)原則》(S9:Nonclinical Evaluation for Anticancer Pharmaceuticals) 及問(wèn)答文件(S9 Implementation Working Group Questions and Answers) 提供了抗腫瘤藥物非臨床評(píng)估的相關(guān)指導(dǎo)和建議。④ ICH《S7A :人用藥品安全藥理學(xué)試驗(yàn)指導(dǎo)原則》(S7A: Safety Pharmacology Studies for Human Pharmaceuticals)提供了探索對(duì)人體核心系統(tǒng)影響的非臨床安全藥理學(xué)的研究策略等[51-53]。
總體而言,臨床前的試驗(yàn)藥物安全性評(píng)估主要包括藥物作用機(jī)制及其作用靶點(diǎn)、安全藥理學(xué)及毒理學(xué)等內(nèi)容,旨在確定試驗(yàn)藥物的靶標(biāo)效應(yīng)和脫靶效應(yīng),識(shí)別潛在的器官毒性,發(fā)掘毒性與藥物暴露之間的關(guān)系,評(píng)估這些毒性效應(yīng)與人體的潛在相關(guān)性,以及鑒定和(或)確認(rèn)可用于臨床檢測(cè)的安全性生物標(biāo)志物等[45]。本共識(shí)著重對(duì)小分子化學(xué)藥物、生物大分子藥物以及預(yù)防性疫苗的Ⅰ期臨床試驗(yàn)常見(jiàn)且重要的安全性風(fēng)險(xiǎn)點(diǎn)進(jìn)行介紹。
3.3.4.1 小分子化學(xué)藥物
小分子化學(xué)藥物在Ⅰ期臨床試驗(yàn)中可能會(huì)給參與者帶來(lái)的重要安全風(fēng)險(xiǎn)包括心臟毒性、肝臟毒性和腎臟毒性。
(1)心臟毒性。在小分子化學(xué)藥物的健康參與者早期臨床試驗(yàn)中,關(guān)注心臟毒性尤為重要,主要包括:① QT 間期延長(zhǎng)。一些非抗心律失常藥物具有使心臟復(fù)極化延遲的不良作用, 在體表心電圖上表現(xiàn)為QT 間期延長(zhǎng)。如果在研究中發(fā)現(xiàn)有臨床意義的結(jié)果,包括但不限于QT 間期較基線(xiàn)的變化,則由研究者或有資質(zhì)的指定人員確定參與者是否可以繼續(xù)接受研究用藥, 以及是否需要進(jìn)行治療。對(duì)心電圖的判讀結(jié)果必須予以記錄。具體內(nèi)容可參考ICH《E14 :非抗心律失常藥物QT/QTc 間期延長(zhǎng)及致心律失常潛力的臨床評(píng)價(jià)》(E14: The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs)[54]。② 心律失常。某些藥物還可以引發(fā)心律失常,包括緩慢型心律失常(如竇性心動(dòng)過(guò)緩、房室傳導(dǎo)阻滯)、心房撲動(dòng)、心房顫動(dòng)、房室折返性心動(dòng)過(guò)速、單形性室性心動(dòng)過(guò)速和Brugada 綜合征。某些類(lèi)型的心律失常(如緩慢型心律失常、心房撲動(dòng)、房室折返性心動(dòng)過(guò)速)主要因癥狀顯著而被關(guān)注;而另一些類(lèi)型(如單形性室性心動(dòng)過(guò)速、Brugada 綜合征、尖端扭轉(zhuǎn)型室性心動(dòng)過(guò)速)可能導(dǎo)致嚴(yán)重后果,乃至猝死。雖然某些藥物引發(fā)心律失常的作用機(jī)制已較為明確,但仍有很多創(chuàng)新藥物的致病機(jī)制尚不清楚,因此加強(qiáng)心電圖檢測(cè)和其他相關(guān)監(jiān)測(cè)策略對(duì)于早期發(fā)現(xiàn)和治療這類(lèi)情況具有重要意義。③心肌和心臟瓣膜損傷。目前已有多種藥物被發(fā)現(xiàn)可直接影響心臟功能,導(dǎo)致直接的心肌組織毒性或瓣膜疾病,例如,蒽環(huán)類(lèi)抗生素和酪氨酸激酶抑制劑可能引起可逆的劑量依賴(lài)性心肌功能減退,進(jìn)而導(dǎo)致心力衰竭。這些效應(yīng)可能會(huì)受到同步進(jìn)行的放射治療或與其他藥物(如甲氨蝶呤或紫杉醇)聯(lián)合治療的影響。對(duì)于具有潛在心肌組織毒性或瓣膜損害的藥物,在臨床試驗(yàn)中應(yīng)加強(qiáng)對(duì)心電圖和生命體征的監(jiān)測(cè),尤其是超聲心動(dòng)圖檢查和心肌標(biāo)記物的測(cè)定。④血壓影響。部分藥物可能通過(guò)影響鈉平衡、上調(diào)腎上腺素水平或抑制副交感神經(jīng)活性,影響腎素- 血管緊張素系統(tǒng)的調(diào)節(jié)功能,或?qū)?nèi)皮細(xì)胞或血管平滑肌產(chǎn)生直接作用等影響血壓。某些藥物引起的高血壓可能導(dǎo)致嚴(yán)重的并發(fā)癥,如動(dòng)脈瘤、腎功能衰竭、心臟病發(fā)作、心力衰竭或卒中。直立性低血壓是多種藥物的常見(jiàn)不良反應(yīng)之一,表現(xiàn)為站立時(shí)血壓明顯降低,腦部血流供應(yīng)減少,與跌倒風(fēng)險(xiǎn)增加、卒中、認(rèn)知功能障礙和死亡率上升等密切相關(guān)。據(jù)統(tǒng)計(jì),有超過(guò)250種藥物可誘發(fā)直立性低血壓[55]。在嚴(yán)重情況下,直立性低血壓可能導(dǎo)致暈厥、意外跌倒,或使重要器官因缺血而受損。因此當(dāng)已知研究藥物可能涉及相關(guān)作用機(jī)制時(shí),應(yīng)在試驗(yàn)方案中明確血壓監(jiān)測(cè)方案,以保護(hù)參與者安全。
(2)肝臟毒性。藥物性肝損傷(drug-induced liver injury,DILI)是指使用藥物(包括處方藥、非處方藥)、中藥材等產(chǎn)品后出現(xiàn)的肝臟功能損傷。DILI 是藥物臨床研發(fā)失敗的重要安全性因素之一。通常情況下,導(dǎo)致嚴(yán)重 DILI 的肝損傷類(lèi)型主要是肝細(xì)胞損傷,其特征在于血清中轉(zhuǎn)氨酶活性顯著升高, 具體表現(xiàn)為丙氨酸氨基轉(zhuǎn)移酶(alanine aminotransferase,ALT) 或天冬氨酸氨基轉(zhuǎn)移酶(aspartate transferase,AST) 從損傷的肝細(xì)胞中釋放至血液。然而,藥物引發(fā)某些肝細(xì)胞損傷的能力并不能準(zhǔn)確預(yù)示其是否有可能導(dǎo)致嚴(yán)重的肝臟損害綜合征。部分藥物雖然會(huì)導(dǎo)致血清轉(zhuǎn)氨酶活性的短暫升高,但持續(xù)用藥并不必然導(dǎo)致進(jìn)行性或嚴(yán)重的 DILI。只有那些能廣泛損傷肝細(xì)胞,進(jìn)而影響肝臟清除血漿膽紅素或合成凝血酶原及其他凝血因子功能的藥物,才可能誘發(fā)嚴(yán)重的 DILI。當(dāng)出現(xiàn)急性肝細(xì)胞損傷時(shí),需立刻停用導(dǎo)致肝損傷的相關(guān)藥物,并采取相應(yīng)的治療措施。
目前可用于預(yù)測(cè)DILI 轉(zhuǎn)歸的生物標(biāo)志物十分有限。海氏法則是最常用的DILI 判定方法,其定義如下:①藥物引起的肝損傷,通常表現(xiàn)為與無(wú)肝毒性的對(duì)照藥物或安慰劑相比,試驗(yàn)組中較高比例的病例出現(xiàn)ALT 或AST 水平升高至3 倍正常值上限(upperlimit of normal,ULN)或以上。② 在ALT 或AST 水平升高的參與者中,某些患者還會(huì)伴有轉(zhuǎn)氨酶水平高于3×ULN,同時(shí)血清總膽紅素升高超過(guò)2×ULN,且血清堿性磷酸酶活性不超過(guò)2×ULN。③無(wú)其他原因可以解釋轉(zhuǎn)氨酶水平和總膽紅素水平的聯(lián)合升高,例如,已經(jīng)排除病毒性甲型肝炎、乙型肝炎或丙型肝炎,先前已存在的或急性肝病,或能引起所觀察到的肝損傷的其他藥物。
在研究方案中應(yīng)當(dāng)規(guī)定肝臟生化檢查異常需要停止用藥的標(biāo)準(zhǔn),以保護(hù)參與者安全。DILI 診斷是排除性診斷,全面、細(xì)致地追溯可疑用藥史和除外其他肝損傷的病因?qū)υ\斷至關(guān)重要。具體可參考2023 年7 月藥審中心發(fā)布的《臨床試驗(yàn)中的藥物性肝損傷識(shí)別、處理及評(píng)價(jià)指導(dǎo)原則》[56]。
(3)腎臟毒性。腎臟在藥物排泄方面具有關(guān)鍵作用,腎臟的高過(guò)濾能力和代謝活性特征使其易受藥物損傷,因此深入理解腎臟生理特性及病理變化機(jī)制對(duì)評(píng)估藥物安全性至關(guān)重要。藥物引起的腎毒性可能是多種因素綜合作用的結(jié)果,包括試驗(yàn)藥物的作用機(jī)制(靶內(nèi)效應(yīng)和脫靶效應(yīng))、特定的研究人群(如老年人、多囊腎患者)、開(kāi)始用藥時(shí)已有腎功能損害者(此處特指腎功能減退PK 研究中的研究對(duì)象)、個(gè)體遺傳傾向和(或)伴隨用藥。不同藥物的腎毒性表現(xiàn)存在很大差異,部分癥狀可能在接觸某些藥物(如造影劑)后立即出現(xiàn);也可能在數(shù)天至數(shù)周后出現(xiàn),如接觸非甾體抗炎藥或氨基糖苷類(lèi)抗生素;或僅在長(zhǎng)期接觸違規(guī)藥物后出現(xiàn),如苯乙雙胍或環(huán)孢素。部分一過(guò)性和輕微程度的藥源性腎病(drug-induced nephropathy,DIN),可能因沒(méi)有癥狀或癥狀較輕微而易被忽略。因此應(yīng)在具有相關(guān)風(fēng)險(xiǎn)的藥物研究方案中完善關(guān)于腎功能評(píng)估的相關(guān)內(nèi)容,以識(shí)別早期和延遲發(fā)生的 DIN。
3.3.4.2 生物大分子藥物
生物大分子藥物(包括單克隆抗體和其他治療性蛋白制劑等)與小分子化學(xué)藥物的主要區(qū)別在于分子大小、結(jié)構(gòu)復(fù)雜性和生產(chǎn)方式[57]。生物大分子藥物本身具有生物活性,因此其毒性作用通常與藥效相關(guān),且脫靶效應(yīng)較為少見(jiàn)。生物大分子藥物常見(jiàn)的AE 有免疫原性、免疫相關(guān)不良事件(immune-related adverse event,irAE)、輸液相關(guān)反應(yīng)(infusion related reaction,IRR)、注射部位反應(yīng)(injectionsite reactions,ISR)及其他[21]。
(1)免疫原性。藥物的免疫原性是指藥物和(或)其代謝物誘發(fā)對(duì)自身或相關(guān)蛋白的免疫應(yīng)答或免疫相關(guān)事件的能力[58]。具有免疫原性的藥物可能誘發(fā)機(jī)體產(chǎn)生不必要的免疫反應(yīng),產(chǎn)生抗藥抗體(anti-drug antibody,ADA)。ADA 可通過(guò)T 細(xì)胞依賴(lài)和T 細(xì)胞非依賴(lài)兩種途徑產(chǎn)生, 其中包括B 細(xì)胞在T 細(xì)胞輔助和不輔助下產(chǎn)生抗體。ADA包括藥物結(jié)合抗體(binding antibodies,BAbs)和中和抗體( neutralizing antibodies ,NAbs)[59], 其中BAbs 會(huì)引起嚴(yán)重危及生命的免疫反應(yīng), 而NAbs 具備藥物中和能力,能夠抑制藥物的生物活性以降低藥效。
藥物的免疫原性影響廣泛,對(duì)參與者的安全和藥物有效性均有重要影響。其中,過(guò)敏反應(yīng)是最常導(dǎo)致的安全性問(wèn)題。
(2)免疫相關(guān)不良事件。免疫治療藥物的毒性和不良反應(yīng)多數(shù)由正常器官的過(guò)度免疫應(yīng)答引起。irAE 的發(fā)生機(jī)制可能包括:① T 細(xì)胞對(duì)腫瘤細(xì)胞和正常組織共同攜帶抗原的活性增強(qiáng)。②已存在的自身抗體水平升高。③炎癥細(xì)胞因子水平升高。④補(bǔ)體介導(dǎo)的炎癥反應(yīng)增強(qiáng),同時(shí)可誘發(fā)T 細(xì)胞和正常細(xì)胞上抗原間的交叉反應(yīng)和自身抗體的產(chǎn)生[60]。
目前,irAE 的發(fā)生率為15%~90%[61],通常于免疫治療開(kāi)始后的數(shù)周至數(shù)月內(nèi)發(fā)生。irAE 幾乎可累及所有器官/ 系統(tǒng),最常發(fā)生的器官/ 系統(tǒng)疾病及主要臨床表現(xiàn)包括:皮膚及皮膚附件疾病,如皮疹、皮炎等;胃腸疾病,如腸炎等;肝膽疾病,如肝炎、肝酶升高等;內(nèi)分泌疾病,如甲狀腺功能減退癥、甲狀腺功能亢進(jìn)、垂體疾病等[62]。
此外,免疫治療藥物,尤其是淋巴細(xì)胞相關(guān)性抗體, 還可能引起相對(duì)少見(jiàn)的細(xì)胞因子釋放綜合征(cytokine release syndrome,CRS), 即一種發(fā)生于免疫治療后的、可能危及生命的超生理反應(yīng),可導(dǎo)致內(nèi)源性或輸注的T 細(xì)胞和(或)其他免疫效應(yīng)細(xì)胞被激活或參與。CRS的癥狀可能為漸進(jìn)性的,發(fā)病時(shí)一定包括發(fā)熱,可能包括低血壓、毛細(xì)血管滲漏綜合征(缺氧)和終末器官功能障礙。CRS 的特征性表現(xiàn)為用藥后1~2 h 出現(xiàn)腫瘤壞死因子-α、干擾素-γ、白介素(interleukin,IL)-2、IL-6血清水平升高,部分病例還可見(jiàn)IL-4、IL-5、IL-8、IL-10、IL-12、IL-13、IL-17、IL-22 和補(bǔ)體水平升高,實(shí)驗(yàn)室檢查中升高的指標(biāo)還有ALT、AST、C 反應(yīng)蛋白、血肌酐、D- 二聚體、γ- 谷氨酰轉(zhuǎn)移酶、乳酸脫氫酶、血磷、尿酸,下降的指標(biāo)有血鈣、血漿蛋白、血紅蛋白、血小板和白細(xì)胞等。因此監(jiān)測(cè)上述實(shí)驗(yàn)室檢查指標(biāo)可能有助于早期識(shí)別CRS [63]。
(3)輸液相關(guān)反應(yīng)。IRR 是由輸注藥物或生物學(xué)成分引起的不良反應(yīng),其發(fā)生機(jī)制分為2 類(lèi),一類(lèi)為過(guò)敏反應(yīng),一般由免疫球蛋白E 介導(dǎo);另一類(lèi)則是非典型的過(guò)敏反應(yīng),稱(chēng)為類(lèi)過(guò)敏反應(yīng),例如,某些細(xì)胞因子釋放產(chǎn)生的反應(yīng)。IRR 相關(guān)的癥狀通常表現(xiàn)為低熱、寒戰(zhàn)、頭痛或惡心,嚴(yán)重者可能出現(xiàn)心動(dòng)過(guò)速、血壓不穩(wěn)定、低氧血癥、胸痛、咳嗽、呼吸急促、喘息、潮紅、出汗、蕁麻疹或瘙癢、血管性水腫、先兆暈厥或暈厥等其他癥狀[64]。
(4)注射部位反應(yīng)。ISR 通常是速發(fā)型的或在注射后1~2 天出現(xiàn),之后常隨著時(shí)間的推移逐漸減弱。臨床表現(xiàn)為注射部位瘙癢、紅斑和水腫等。通常認(rèn)為注射部位反應(yīng)也是一種由T 細(xì)胞介導(dǎo)的超敏反應(yīng),其中CD8+T 細(xì)胞為主要效應(yīng)細(xì)胞,但也可能涉及其他機(jī)制[65]。
(5)其他。生物大分子藥物分子量較大,可造成腎臟排泄困難,進(jìn)而容易引起腎臟炎癥反[57]。例如, 免疫檢查點(diǎn)抑制劑( 如PD-1)因其藥理學(xué)和免疫調(diào)節(jié)特性,在約2% 的患者群體中,可能與急性腎損傷和(或)急性間質(zhì)性腎炎的發(fā)生相關(guān)[66]。
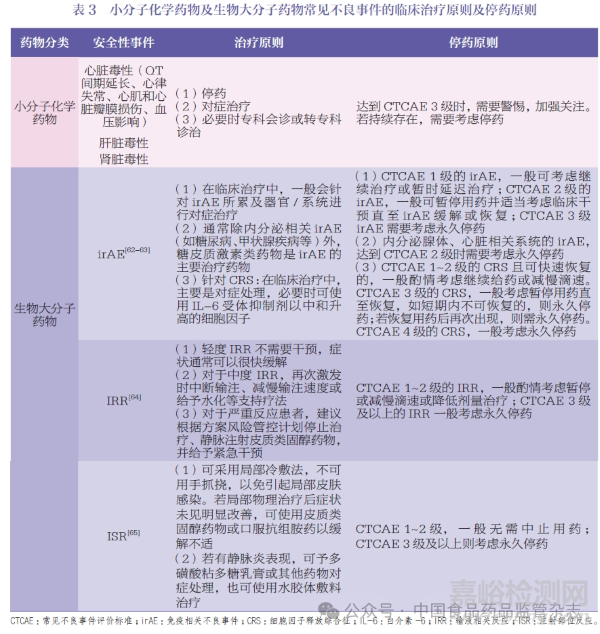
一般相關(guān)AE 的處理原則和治療方案均會(huì)體現(xiàn)在臨床試驗(yàn)方案中,通常會(huì)結(jié)合AE的嚴(yán)重程度、累及器官/ 系統(tǒng)以及持續(xù)時(shí)間來(lái)綜合考慮,本共識(shí)簡(jiǎn)要匯總了小分子化學(xué)藥物和生物大分子藥物常見(jiàn)AE 的臨床治療及停藥原則,見(jiàn)表3[62-65]。
3.3.4.3 預(yù)防性疫苗
預(yù)防性疫苗通常也為生物制品,若其來(lái)源于活生物體,則成分通常較為復(fù)雜。預(yù)防性疫苗常用于健康人群,且以?xún)和癁橹饕臃N對(duì)象,因此在安全性和有效性方面具有特殊性。通常,Ⅰ期臨床試驗(yàn)重點(diǎn)觀察安全性,觀察對(duì)象首先應(yīng)為健康成人,并按照從成人到兒童、再到嬰幼兒的順序逐步開(kāi)展臨床試驗(yàn)。
疫苗臨床試驗(yàn)將AE 分為征集性AE 和非征集性AE,其中征集性AE 又分為局部征集性AE和全身征集性AE。常見(jiàn)局部征集性AE 包括接種部位疼痛、觸痛、硬結(jié)、腫脹,皮疹、紅暈、瘙癢、蜂窩組織炎等;常見(jiàn)全身征集性AE 累及器官/ 系統(tǒng)(癥狀/ 體征)包括:胃腸系統(tǒng)(如腹瀉、便秘、吞咽困難、厭食、惡心、嘔吐)、肌肉骨骼與結(jié)締組織(如非接種部位肌肉痛、關(guān)節(jié)炎、關(guān)節(jié)痛)、神經(jīng)系統(tǒng)(如頭痛、暈厥、新發(fā)驚厥)、呼吸系統(tǒng)(如咳嗽、急性支氣管痙攣、呼吸困難)、皮膚及皮下組織(如非接種部位瘙癢且無(wú)皮膚損傷、皮膚黏膜異常)、精神系統(tǒng)(如失眠、激惹或抑制、精神障礙)、免疫系統(tǒng)(如急性過(guò)敏反應(yīng))以及其他(如發(fā)熱、疲勞、乏力、非接種部位疼痛)等。征集性AE 的具體定義和內(nèi)容會(huì)因不同疫苗特性而存在差異,具體需根據(jù)方案執(zhí)行。所有AE 均應(yīng)按照2019 年國(guó)家藥監(jiān)局發(fā)布的《預(yù)防用疫苗臨床試驗(yàn)不良事件分級(jí)標(biāo)準(zhǔn)指導(dǎo)原則》[48] 中的5 級(jí)分類(lèi)標(biāo)準(zhǔn)進(jìn)行評(píng)價(jià)。
此外,不同類(lèi)型疫苗在安全性評(píng)價(jià)方面有一些特殊考量:①減毒活疫苗(病毒或細(xì)菌)可能在受種者及其接觸者中造成嚴(yán)重感染,除常規(guī)安全性評(píng)價(jià)外,還應(yīng)盡早考慮排毒、接觸傳播、遺傳穩(wěn)定性和毒力返祖等方面。對(duì)于排毒的監(jiān)測(cè)需基于病原體和疾病特征、疫苗特性及接種途徑,并結(jié)合臨床前研究和同類(lèi)已上市疫苗的研究數(shù)據(jù)進(jìn)行科學(xué)合理的設(shè)計(jì),包括明確研究目的、評(píng)價(jià)指標(biāo)、樣本類(lèi)型、病原體檢測(cè)時(shí)間及方法等。②病毒載體類(lèi)疫苗應(yīng)關(guān)注載體病毒對(duì)人體的影響,同時(shí)考慮受試者體內(nèi)預(yù)存抗體、是否再?gòu)?fù)制等。③核酸類(lèi)疫苗應(yīng)根據(jù)其制劑組成、結(jié)構(gòu)和工藝等特點(diǎn),如新型遞送系統(tǒng)/ 新輔料的使用、體內(nèi)生物分布及存續(xù)時(shí)間等,關(guān)注相應(yīng)的安全性風(fēng)險(xiǎn)。
3.3.5 AE 記錄中的爭(zhēng)議問(wèn)題及建議
在臨床試驗(yàn)中,AE 是評(píng)估試驗(yàn)藥物安全性的重要指標(biāo)。為確保AE 的統(tǒng)一記錄和報(bào)告,AE命名應(yīng)盡量使用標(biāo)準(zhǔn)醫(yī)學(xué)術(shù)語(yǔ),建議參照《監(jiān)管活動(dòng)醫(yī)學(xué)詞典》(Medical Dictionary for Regulatory Activities,MedDRA)的首選術(shù)語(yǔ)(preferred terms,PT) 或低位術(shù)語(yǔ)(lowest level term,LLT)進(jìn)行,以提高數(shù)據(jù)的一致性和可比性。此外,AE 記錄也應(yīng)完整,需包含發(fā)生時(shí)間、事件描述、嚴(yán)重程度、與試驗(yàn)藥物的相關(guān)性、采取的措施、轉(zhuǎn)歸或結(jié)果,以符合核查要求。記錄AE 時(shí),應(yīng)首先記錄診斷而不是癥狀,只有在不能做出診斷時(shí)記錄癥狀、體征或?qū)嶒?yàn)室檢查指標(biāo)異常。當(dāng)后期診斷明確時(shí),應(yīng)對(duì)記錄進(jìn)行更新,即以明確的診斷代替之前的癥狀、體征或?qū)嶒?yàn)室檢查指標(biāo)異常。在AE 記錄中,對(duì)于一些常見(jiàn)分歧和爭(zhēng)議,本共識(shí)提出相關(guān)建議,具體內(nèi)容如下。
爭(zhēng)議一:AE 開(kāi)始和結(jié)束時(shí)間是否需要具體到分鐘。在一些較長(zhǎng)周期的臨床試驗(yàn)隨訪(fǎng)中,有時(shí)參與者僅能提供AE 發(fā)生的日期,卻無(wú)法提供時(shí)間點(diǎn)。因此為了盡可能完整地收集原始數(shù)據(jù),并保證原始數(shù)據(jù)的準(zhǔn)確性和可靠性,本共識(shí)建議:參與者在入住研究中心期間,因每日均有研究醫(yī)生查房問(wèn)詢(xún)AE,故而AE 開(kāi)始和結(jié)束時(shí)間應(yīng)盡量具體到分鐘。出院后若隨訪(fǎng)時(shí)間間隔較久,無(wú)法問(wèn)詢(xún)到AE 具體時(shí)間點(diǎn),則如實(shí)記錄到日期,具體時(shí)間點(diǎn)記為不詳即可。另外,研究者也要加強(qiáng)對(duì)參與者的宣教和管理,告知參與者有任何不適,應(yīng)查看并記錄發(fā)生的日期及具體時(shí)間,并第一時(shí)間告知研究者,以便研究者跟進(jìn)評(píng)估AE 的嚴(yán)重程度并指導(dǎo)后續(xù)治療。
爭(zhēng)議二:AE 嚴(yán)重程度升級(jí)后,是記錄為1 條新的AE,還是按照最高級(jí)別記錄。對(duì)于嚴(yán)重程度升級(jí)的AE,目前常見(jiàn)的記錄方式有2 種,一種是記錄為1 條新的AE,另一種是全程僅記錄1 條AE,級(jí)別按照最高級(jí)記錄。具體選擇哪種方式,應(yīng)該在項(xiàng)目啟動(dòng)前盡早與申辦者溝通確認(rèn),同一試驗(yàn)或類(lèi)似試驗(yàn)中應(yīng)使用相同的標(biāo)準(zhǔn)。本共識(shí)推薦全程記錄1 條AE,按最高級(jí)別記錄嚴(yán)重程度。
爭(zhēng)議三:反復(fù)發(fā)生的AE,是按照發(fā)生頻次記錄多條,還是只記為1 條。結(jié)合行業(yè)內(nèi)共識(shí)[67],本共識(shí)推薦:對(duì)于一段時(shí)間內(nèi)反復(fù)發(fā)生的AE,如果前后是有關(guān)聯(lián)的(屬于之前AE 的持續(xù)、進(jìn)展或者復(fù)發(fā)),建議作為同一AE 在病歷中進(jìn)行記錄,并結(jié)合之前的記錄對(duì)嚴(yán)重程度進(jìn)行說(shuō)明(如藥物導(dǎo)致的手足皮膚反應(yīng),可持續(xù)存在,但時(shí)輕時(shí)重);如果前后并無(wú)關(guān)聯(lián)(如研究不同階段出現(xiàn)的兩次肺部感染),則分別作為單獨(dú)的AE 在病歷中記錄。當(dāng)難以判斷關(guān)聯(lián)性時(shí),也可協(xié)商一個(gè)間隔時(shí)間(如2 周)來(lái)規(guī)定記錄次數(shù)。
爭(zhēng)議四:在某一實(shí)驗(yàn)室檢查項(xiàng)目中,多個(gè)指標(biāo)結(jié)果異常,可否合并記錄為1 條AE。以血常規(guī)檢查結(jié)果為例,若白細(xì)胞計(jì)數(shù)和中性粒細(xì)胞絕對(duì)值均降低或均升高,是合并記錄為1 條AE,還是分開(kāi)記錄為2 條;若中性粒細(xì)胞百分比和絕對(duì)值均降低或均升高,是記錄為1 條AE,還是分開(kāi)記錄為2 條。針對(duì)此類(lèi)情況,本共識(shí)建議按照臨床意義區(qū)分。
情況1 :如僅僅只是白細(xì)胞計(jì)數(shù)降低,合并中性粒細(xì)胞或淋巴細(xì)胞絕對(duì)值降低,無(wú)臨床癥狀或體征,考慮白細(xì)胞計(jì)數(shù)降低為中性粒細(xì)胞或淋巴細(xì)胞降低引起,則僅記錄中性粒細(xì)胞或淋巴細(xì)胞絕對(duì)值降低;如果難以明確降低原因,可根據(jù)臨床判斷分開(kāi)報(bào)告;如果符合粒細(xì)胞缺乏等嚴(yán)重的臨床診斷標(biāo)準(zhǔn),需按照診斷記錄。對(duì)于白細(xì)胞計(jì)數(shù)和中性粒細(xì)胞絕對(duì)值均升高的情形,可采用同樣的方式分類(lèi)記錄。
情況2 :中性粒細(xì)胞絕對(duì)值和百分比同時(shí)出現(xiàn)升高或降低,若絕對(duì)值的變化具有臨床意義,建議僅記錄該指標(biāo)的絕對(duì)值。對(duì)于淋巴細(xì)胞、單核細(xì)胞、嗜酸性粒細(xì)胞、嗜堿性粒細(xì)胞絕對(duì)值和百分比同步變化的情形,可采用同樣的處理方式。
爭(zhēng)議五:皮膚外用藥給藥部位AE,按照接觸性皮炎的診斷名稱(chēng)進(jìn)行記錄,還是按照每個(gè)癥狀和(或)體征單獨(dú)記錄。皮膚外用藥可能會(huì)導(dǎo)致皮膚刺激性和(或)過(guò)敏性等局部不良反應(yīng),表現(xiàn)為皮膚紅斑、丘疹、瘙癢、疼痛、腫脹、滲出等。常見(jiàn)的是接觸性皮炎,其他少見(jiàn)反應(yīng)有色素改變、脫毛等。外用藥皮膚局部不良反應(yīng)并不少見(jiàn),針對(duì)接觸性皮炎,國(guó)內(nèi)外均制定了評(píng)分標(biāo)準(zhǔn)。接觸性皮炎又分為刺激性皮炎、變應(yīng)性接觸性皮炎、速發(fā)型接觸性反應(yīng)、光毒性及光變應(yīng)性接觸性皮炎,不同分型之間的鑒別也存在一定難度[68-69]。因此對(duì)于參與者使用皮膚外用藥后出現(xiàn)的局部體征,必要時(shí)需皮膚科專(zhuān)科醫(yī)生介入,協(xié)助診斷及分型,以指導(dǎo)后續(xù)的處理措施。建議在記錄AE時(shí),若符合疾病診斷,則按照診斷進(jìn)行記錄,以更好地理解藥物不良反應(yīng)的發(fā)生機(jī)制;若僅為體征,難以診斷為某一類(lèi)疾病,則根據(jù)單獨(dú)的體征進(jìn)行記錄,但也需要參照MedDRA 的PT 或LLT 進(jìn)行命名。
3.4 方案設(shè)計(jì)中的安全性風(fēng)險(xiǎn)管理
為有效降低參與者風(fēng)險(xiǎn),Ⅰ期臨床試驗(yàn)應(yīng)制定科學(xué)且可行的風(fēng)險(xiǎn)評(píng)估和控制計(jì)劃。在方案設(shè)計(jì)層面,需要充分考慮科學(xué)有效的安全監(jiān)測(cè)和藥物風(fēng)險(xiǎn)管理策略。方案制定是多方參與的過(guò)程,由申辦者主導(dǎo)和負(fù)責(zé)、研究者參與討論、倫理委員會(huì)審核批準(zhǔn)。在臨床試驗(yàn)方案中需要對(duì)藥物的安全性特性,特別是其重要風(fēng)險(xiǎn),進(jìn)行詳細(xì)闡述,充分評(píng)估藥物對(duì)于參與者的風(fēng)險(xiǎn)和獲益,并在此基礎(chǔ)上確定風(fēng)險(xiǎn)控制措施,以保證參與者的安全。
藥物研發(fā)是漸進(jìn)式探索的過(guò)程,涉及對(duì)動(dòng)物和人體有效性和安全性等的持續(xù)評(píng)價(jià)。非臨床安全性評(píng)價(jià)的信息可用于估算人體臨床試驗(yàn)的安全起始劑量和劑量范圍,確定潛在不良反應(yīng)的臨床監(jiān)測(cè)指標(biāo)。此外,相同或類(lèi)似機(jī)制藥物的臨床使用經(jīng)驗(yàn)和安全信息也是制定首次人體試驗(yàn)風(fēng)險(xiǎn)管理策略的重要參考信息。開(kāi)展任何臨床試驗(yàn)之前,其非臨床研究或以往臨床研究的結(jié)果必須足以說(shuō)明藥物在所推薦的人體研究中有可接受的安全性基礎(chǔ)。在整個(gè)藥物研發(fā)過(guò)程中,應(yīng)當(dāng)動(dòng)態(tài)地對(duì)藥理、毒理等臨床數(shù)據(jù)進(jìn)行評(píng)價(jià),以評(píng)估臨床試驗(yàn)可能給參與者帶來(lái)的安全性風(fēng)險(xiǎn)。對(duì)于正在或?qū)⒁M(jìn)行的臨床試驗(yàn)方案,也應(yīng)進(jìn)行必要的調(diào)整[70]。針對(duì)Ⅰ期臨床試驗(yàn)方案中的參與者安全管理,本共識(shí)建議考慮以下幾個(gè)方面。
3.4.1 參與者入選和排除標(biāo)準(zhǔn)
對(duì)于首次人體試驗(yàn),健康參與者并不會(huì)從中獲得臨床益處,因此在方案中需盡量全面闡述可能導(dǎo)致安全風(fēng)險(xiǎn)增加的因素,并設(shè)定明確詳細(xì)的入選和排除標(biāo)準(zhǔn)。根據(jù)不同作用機(jī)制和靶器官毒性等臨床前發(fā)現(xiàn),充分考量可能增加健康參與者風(fēng)險(xiǎn)的既往病史、用藥情況、治療方案以及實(shí)驗(yàn)室檢查結(jié)果等,確保試驗(yàn)對(duì)象的臨床特征符合研究目的,避免可能影響藥物效果或增加風(fēng)險(xiǎn)的基礎(chǔ)疾病或合并用藥,從而最大程度控制潛在的風(fēng)險(xiǎn)。
3.4.2 藥物劑量的合理選擇
人體起始劑量的估算是保障首次人體試驗(yàn)參與者安全的一個(gè)重要因素。人體耐受性臨床試驗(yàn)最大推薦起始劑量(maximum recommended starting dose,MRSD)的確定需要參考相關(guān)指導(dǎo)原則和技術(shù)指南(包括定量藥理學(xué)研究方法等)。劑量選擇應(yīng)充分考慮藥物的藥效學(xué)特性,包括藥物的最小有效劑量、最大耐受劑量等,以確保選擇的劑量范圍能夠在試驗(yàn)中展現(xiàn)出藥物的預(yù)期研究目的,同時(shí)避免患者受到超劑量藥物的毒性影響。通常,在最適合的動(dòng)物種屬中進(jìn)行的非臨床安全性試驗(yàn)確定的未見(jiàn)明顯毒性反應(yīng)劑量(no observed adverseeffect level,NOAEL) 可提供最重要的參考信息。藥代動(dòng)力學(xué)/ 藥效學(xué)(pharmacokinetic/pharmacodynamic,PK/PD)建模也在劑量選擇時(shí)被廣泛使用,通過(guò)定量框架模擬和幫助選擇治療劑量范圍。首次人體試驗(yàn)的藥物劑量一般從最低單一劑量開(kāi)始。為進(jìn)一步降低首次人體試驗(yàn)的風(fēng)險(xiǎn),特別是當(dāng)藥物存在潛在嚴(yán)重風(fēng)險(xiǎn)或不確定性較高時(shí),在試驗(yàn)早期進(jìn)行劑量遞增時(shí)可采用哨兵給藥方式,即在組內(nèi)其他參與者給藥前,選取1~2 例參與者給予單一劑量的試驗(yàn)藥物,該參與者與組內(nèi)其他受試者應(yīng)間隔充足的觀察期以便觀察任何可能的AE。在充分評(píng)估單一劑量試驗(yàn)結(jié)果(包括PK/PD 、安全性數(shù)據(jù)等)后,確保受試者安全的情況下,進(jìn)一步進(jìn)行多劑量和遞增給藥的研究。劑量遞增是一個(gè)動(dòng)態(tài)調(diào)整的過(guò)程,應(yīng)確保及時(shí)對(duì)已有的非臨床和(或)臨床數(shù)據(jù)進(jìn)行分析評(píng)估,必要時(shí)對(duì)后續(xù)遞增劑量進(jìn)行調(diào)整,以降低風(fēng)險(xiǎn)或提高遞增效率。試驗(yàn)方案設(shè)計(jì)中必須明確闡述終止標(biāo)準(zhǔn),即出現(xiàn)哪些不良事件或者達(dá)到何種暴露量,如峰濃度(peakconcentration,Cmax)或曲線(xiàn)下面積(area under the curve,AUC)時(shí),劑量遞增試驗(yàn)應(yīng)終止等。
3.4.3 監(jiān)測(cè)和評(píng)估機(jī)制的具體規(guī)定
在臨床試驗(yàn)過(guò)程中,需要制定持續(xù)的安全監(jiān)測(cè)計(jì)劃。對(duì)于首次人體試驗(yàn), 方案中需要詳細(xì)描述監(jiān)測(cè)的方法、時(shí)間點(diǎn)和頻率、監(jiān)測(cè)結(jié)果的評(píng)價(jià)標(biāo)準(zhǔn)等,確保能及時(shí)發(fā)現(xiàn)并處理任何可能的安全性風(fēng)險(xiǎn)。同時(shí),為評(píng)價(jià)藥物長(zhǎng)期安全性或及時(shí)發(fā)現(xiàn)遲發(fā)性AE,依據(jù)試驗(yàn)藥物的PK/PD 特征以及臨床研究目的,試驗(yàn)方案中需要設(shè)定合理的隨訪(fǎng)方式和時(shí)間。在首次人體試驗(yàn)中,由于受樣本量和受試人群等限制,只會(huì)觀察到部分或常見(jiàn)AE。臨床試驗(yàn)方案中需描述特別關(guān)注不良事件(adverse event of special interest,AESI),包括此類(lèi)事件的范圍、評(píng)估、處理和報(bào)告標(biāo)準(zhǔn)。在試驗(yàn)進(jìn)行期間一旦出現(xiàn)AESI,需要持續(xù)監(jiān)測(cè)并由研究者與申辦者快速溝通且開(kāi)展相關(guān)研究,以進(jìn)一步了解藥物安全特征。在首次人體試驗(yàn)開(kāi)始前,AESI 的確定通常基于試驗(yàn)藥物臨床前、已完成臨床試驗(yàn)(如有)或同靶點(diǎn)藥物安全性信息等。對(duì)于生物制品,動(dòng)物實(shí)驗(yàn)研究通常不能準(zhǔn)確預(yù)測(cè)人類(lèi)的免疫反應(yīng),所以首次人體試驗(yàn)是評(píng)估藥物免疫原性的第一關(guān)。免疫原性對(duì)藥物的安全性和療效都可能產(chǎn)生重大影響,因此需要及時(shí)監(jiān)測(cè)和評(píng)估相關(guān)AE。此外,必要時(shí)申辦方也可以設(shè)立一個(gè)獨(dú)立的安全監(jiān)查委員會(huì),并在方案中描述其主要職責(zé)和工作機(jī)制。
3.4.4 藥物相互作用的管理
基于藥物作用機(jī)制、PK 特征以及非臨床研究結(jié)果,在試驗(yàn)方案中應(yīng)描述和討論所有已知信息和可能的DDI 或食物影響,并在入選和排除標(biāo)準(zhǔn)、用藥方法等章節(jié)加以明確。通常情況下,確認(rèn)相同劑量水平的安全性后,DDI臨床試驗(yàn)或食物影響相關(guān)研究也會(huì)在早期臨床研究階段的健康參與者中進(jìn)行。在設(shè)計(jì)試驗(yàn)方案時(shí),需要充分考慮藥物代謝酶的誘導(dǎo)或抑制效應(yīng)、PK 特征等因素,明確可能因DDI 或食物影響而出現(xiàn)的不良反應(yīng),并給出相應(yīng)的處理措施。
3.4.5 個(gè)體參與者停止標(biāo)準(zhǔn)和試驗(yàn)終止標(biāo)準(zhǔn)
臨床試驗(yàn)方案需明確個(gè)別參與者、部分試驗(yàn)或劑量組或者全部試驗(yàn)的停止標(biāo)準(zhǔn)并嚴(yán)格執(zhí)行。對(duì)于健康參與者的停藥標(biāo)準(zhǔn),一般基于藥物耐受性、PK/PD 特征的評(píng)價(jià)指標(biāo),例如,出現(xiàn)1 例及以上參與者發(fā)生可能與試驗(yàn)藥物相關(guān)的嚴(yán)重不良反應(yīng),或1/3 及以上的參與者發(fā)生可能與試驗(yàn)藥物相關(guān)的CTCAE 2 級(jí)AE, 或藥物暴露量(Cmax 或AUC)超過(guò)預(yù)先設(shè)定的最高標(biāo)準(zhǔn)等。同時(shí),繼續(xù)給藥或臨床試驗(yàn)重啟的標(biāo)準(zhǔn)和流程也需要在方案中予以明確。
3.4.6 揭盲
應(yīng)在試驗(yàn)方案中制定詳細(xì)的緊急揭盲標(biāo)準(zhǔn)操作規(guī)程。臨床試驗(yàn)進(jìn)行過(guò)程中,研究者可以根據(jù)醫(yī)學(xué)判斷或臨床決策提出緊急揭盲的要求,此時(shí)需要及時(shí)采取相應(yīng)措施,確保參與者安全。試驗(yàn)中發(fā)生非預(yù)期嚴(yán)重AE 時(shí),研究者也可對(duì)個(gè)例參與者進(jìn)行緊急揭盲。應(yīng)該通過(guò)合理的設(shè)計(jì)與管理,盡可能保持盲態(tài),從而降低對(duì)臨床試驗(yàn)實(shí)施或最終結(jié)果分析的影響[71]。
3.4.7 方案變更
從非臨床研究階段進(jìn)入臨床研究階段存在高度的不確定性,對(duì)藥物安全風(fēng)險(xiǎn)的認(rèn)識(shí)也是一個(gè)動(dòng)態(tài)過(guò)程。在發(fā)現(xiàn)新的信息并可能影響參與者安全的情況下,方案變更也是風(fēng)險(xiǎn)管理的手段之一。根據(jù)2020 年版《藥品注冊(cè)管理辦法》第二十九條,藥物臨床試驗(yàn)期間,發(fā)生藥物臨床試驗(yàn)方案變更、非臨床或者藥學(xué)的變化或者有新發(fā)現(xiàn)的,申辦者應(yīng)當(dāng)按照規(guī)定,參照相關(guān)技術(shù)指導(dǎo)原則,充分評(píng)估對(duì)受試者安全的影響[72]。對(duì)于臨床試驗(yàn)期間可能增加受試者安全性風(fēng)險(xiǎn)的方案變更,除按規(guī)定向藥審中心提出補(bǔ)充申請(qǐng)外,還應(yīng)嚴(yán)格遵守倫理審查的相關(guān)規(guī)定和要求。
本共識(shí)針對(duì)小分子化學(xué)藥物和生物大分子藥物方案設(shè)計(jì)層面的安全性風(fēng)險(xiǎn)管控要點(diǎn)進(jìn)行了簡(jiǎn)要匯總,主要包括試驗(yàn)藥物安全性風(fēng)險(xiǎn)、藥物靶點(diǎn)或作用機(jī)制、常見(jiàn)藥物以及方案設(shè)計(jì)考量要點(diǎn)等方面內(nèi)容,見(jiàn)表4。

3.5 方案實(shí)施中的安全性風(fēng)險(xiǎn)管控
在Ⅰ期臨床試驗(yàn)中,方案實(shí)施的安全性風(fēng)險(xiǎn)管控直接關(guān)系到參與者的生命安全以及試驗(yàn)的合規(guī)性。在方案實(shí)施階段,建立完善的安全性風(fēng)險(xiǎn)管控體系,是確保參與者權(quán)益、提高試驗(yàn)質(zhì)量和推動(dòng)新藥研發(fā)的重要基礎(chǔ),同時(shí)也是藥物臨床試驗(yàn)機(jī)構(gòu)監(jiān)督檢查的要點(diǎn)內(nèi)容。
3.5.1 制定有效可行的緊急預(yù)案及急救流程
在Ⅰ期臨床試驗(yàn)中,常涉及首次將藥物應(yīng)用于人體,潛在風(fēng)險(xiǎn)較高,項(xiàng)目啟動(dòng)前制定有效的緊急預(yù)案和急救流程是至關(guān)重要的。該預(yù)案需緊密結(jié)合藥物的藥理學(xué)特性、預(yù)期作用、可能的不良反應(yīng)以及特定的試驗(yàn)環(huán)境等,具體應(yīng)包括:①配置緊急醫(yī)療設(shè)備及藥物。如心電監(jiān)護(hù)儀、除顫器、急救藥物等。②定期進(jìn)行專(zhuān)業(yè)培訓(xùn)。對(duì)研究團(tuán)隊(duì),包括醫(yī)生、護(hù)士及其他醫(yī)療相關(guān)人員,定期進(jìn)行急救培訓(xùn),并通過(guò)模擬緊急情況來(lái)檢驗(yàn)流程的有效性。③建立快速響應(yīng)系統(tǒng)。建立一套高效的通信系統(tǒng)及急救流程,確保一旦發(fā)生緊急情況,可以及時(shí)溝通并快速調(diào)動(dòng)有效的醫(yī)療支援。④開(kāi)展全面的風(fēng)險(xiǎn)評(píng)估。基于臨床前安全性評(píng)價(jià)結(jié)果和同類(lèi)藥已知的不良反應(yīng),識(shí)別與藥物相關(guān)的潛在不良反應(yīng)及其嚴(yán)重程度,并預(yù)設(shè)響應(yīng)措施。
3.5.2 建立高效的跨團(tuán)隊(duì)合作與支持機(jī)制
Ⅰ期臨床試驗(yàn)中,為確保有充分的緊急醫(yī)療支持,常涉及Ⅰ期臨床試驗(yàn)研究團(tuán)隊(duì)和多個(gè)臨床科室之間的協(xié)作。高效的合作與支持機(jī)制包含以下要點(diǎn):①明確合作框架和職責(zé)分配。需要建立一個(gè)明確的合作框架,其中詳細(xì)規(guī)定各臨床科室在臨床試驗(yàn)中的角色和職責(zé)。例如,急診科負(fù)責(zé)支持緊急醫(yī)療情況的處理,而相關(guān)專(zhuān)科(如心血管內(nèi)科、神經(jīng)內(nèi)科等)則提供特定條件下的專(zhuān)業(yè)會(huì)診。②建立通信系統(tǒng)。試驗(yàn)期間,及時(shí)溝通與信息共享對(duì)于高效應(yīng)對(duì)突發(fā)事件至關(guān)重要。應(yīng)建立一個(gè)緊急通信協(xié)議,確保在需要時(shí),可以迅速聯(lián)系到相應(yīng)的專(zhuān)科醫(yī)生和急救團(tuán)隊(duì),同時(shí)還應(yīng)明確支援的對(duì)應(yīng)崗位、響應(yīng)時(shí)間、響應(yīng)后到場(chǎng)人員的職責(zé)分工、現(xiàn)場(chǎng)指揮人員及團(tuán)隊(duì)協(xié)作等。③定期開(kāi)展培訓(xùn)與聯(lián)合演練。為了提高各臨床科室間的協(xié)同效率,應(yīng)定期進(jìn)行聯(lián)合培訓(xùn)和應(yīng)急演練。④建立反饋和改進(jìn)機(jī)制。每次緊急情況處理完成后,應(yīng)進(jìn)行詳細(xì)評(píng)估和反饋,包括分析各臨床科室的響應(yīng)時(shí)間、協(xié)作效率以及結(jié)果的有效性等,并根據(jù)反饋結(jié)果及時(shí)調(diào)整和優(yōu)化合作流程及緊急預(yù)案。⑤強(qiáng)化管理支持,保障跨團(tuán)隊(duì)合作。管理層的支持對(duì)于跨團(tuán)隊(duì)合作機(jī)制的成功實(shí)施具有重要作用,同時(shí)也是保障參與者安全的關(guān)鍵。管理層應(yīng)確保各部門(mén)擁有充足的資源,如合格的研究人員、完善的醫(yī)療設(shè)備和充足的資金支持,以滿(mǎn)足臨床試驗(yàn)的安全管理需求。通過(guò)優(yōu)化跨部門(mén)協(xié)作流程、加強(qiáng)信息共享和應(yīng)急響應(yīng)機(jī)制,確保試驗(yàn)過(guò)程中能夠及時(shí)識(shí)別和應(yīng)對(duì)可能影響參與者安全的風(fēng)險(xiǎn)。
3.5.3 安全性審查委員會(huì)決策及風(fēng)險(xiǎn)控制
在包含健康參與者的Ⅰ期臨床試驗(yàn)中, 安全性審查委員會(huì)(Safety Review Committee,SRC)或第三方醫(yī)生負(fù)責(zé)監(jiān)控試驗(yàn)的安全性風(fēng)險(xiǎn)。SRC 通常由具有醫(yī)學(xué)、藥學(xué)、生物統(tǒng)計(jì)學(xué)和臨床研究經(jīng)驗(yàn)的專(zhuān)家組成,獨(dú)立于臨床試驗(yàn)的日常運(yùn)營(yíng),其主要任務(wù)是確保參與者的安全并提供風(fēng)險(xiǎn)控制。
SRC 的主要職責(zé)包括:①定期審查安全性數(shù)據(jù)。SRC 定期接收和審查臨床試驗(yàn)中收集的安全性數(shù)據(jù),包括AE、實(shí)驗(yàn)室檢查結(jié)果以及其他任何有關(guān)參與者健康狀況的報(bào)告。審查這些數(shù)據(jù)有助于及時(shí)識(shí)別潛在的安全問(wèn)題或不良趨勢(shì)。②獲益- 風(fēng)險(xiǎn)評(píng)估。SRC 需要評(píng)估藥物的潛在風(fēng)險(xiǎn)獲益比。特別是在健康人群中進(jìn)行的Ⅰ期臨床試驗(yàn),對(duì)安全性的評(píng)價(jià)應(yīng)尤為嚴(yán)格,原因在于參與者基礎(chǔ)健康狀況良好、所受干擾因素較少,且該階段藥物的治療獲益尚未得到臨床證實(shí)。③提出有關(guān)調(diào)整試驗(yàn)方案的建議。基于安全性數(shù)據(jù)的審查結(jié)果,SRC 可能建議對(duì)試驗(yàn)方案進(jìn)行修改,包括調(diào)整劑量、修改給藥頻率或改變?nèi)脒x和排除標(biāo)準(zhǔn)等,以減少潛在風(fēng)險(xiǎn)。④決定試驗(yàn)暫停或終止。在發(fā)現(xiàn)嚴(yán)重安全問(wèn)題或有數(shù)據(jù)表明繼續(xù)進(jìn)行臨床試驗(yàn)的風(fēng)險(xiǎn)遠(yuǎn)大于潛在益處時(shí),基于多方數(shù)據(jù)支持,SRC 經(jīng)過(guò)仔細(xì)考量后,有權(quán)決定暫停或終止試驗(yàn)。⑤及時(shí)通報(bào)安全信息。SRC 必須確保試驗(yàn)中的所有安全問(wèn)題都能被及時(shí)通報(bào)給所有相關(guān)方,包括研究團(tuán)隊(duì)、倫理委員會(huì)、監(jiān)管機(jī)構(gòu)和參與者。
4. 總結(jié)與展望
總體而言,Ⅰ期臨床試驗(yàn)中健康參與者的安全性管理至關(guān)重要,不僅關(guān)乎參與Ⅰ期臨床試驗(yàn)的參與者本身,也直接影響后續(xù)新藥研發(fā)策略的制定及目標(biāo)適應(yīng)癥患者的用藥安全。本共識(shí)總結(jié)了作者團(tuán)隊(duì)的實(shí)踐經(jīng)驗(yàn)和前沿見(jiàn)解,旨在激發(fā)臨床試驗(yàn)領(lǐng)域的學(xué)術(shù)對(duì)話(huà),推動(dòng)臨床研究學(xué)科發(fā)展,同時(shí)充分保障我國(guó)所有Ⅰ期臨床試驗(yàn)健康參與者的安全與權(quán)益。本共識(shí)內(nèi)容基于臨床疾病相關(guān)指南以及臨床試驗(yàn)相關(guān)指導(dǎo)原則,結(jié)合健康參與者Ⅰ期臨床試驗(yàn)的豐富經(jīng)驗(yàn),經(jīng)綜合評(píng)估與深入討論而形成。鑒于可能存在的疏漏之處,歡迎各位專(zhuān)家和同行提出寶貴意見(jiàn)與指正。
引用本文
楊海靜,趙琳,楊玲,陳銳,張菁*,李海燕*,鄭莉*,韓曉紅*.Ⅰ期臨床試驗(yàn)中健康參與者安全性管理的專(zhuān)家共識(shí)[J].中國(guó)食品藥品監(jiān)管.2025.3(254):44-69.

來(lái)源:中國(guó)食品藥品監(jiān)管雜志


