高分子材料也稱為聚合物材料,是以高分子化合物為基體,再配有其他添加劑(助劑)所構成的材料。高分子材料的結構決定其性能,對結構的控制和改性,可獲得不同特性的高分子材料。高分子材料獨特的結構和易改性、易加工特點,使其具有其他材料不可比擬、不可取代的優異性能,從而廣泛用于科學技術、國防建設和國民經濟各個領域,并已成為現代社會生活中衣食住行用各個方面不可缺少的材料。
主要用于聚合物領域,以有機溶劑為流動相(氯仿,THF,DMF),常用固定相填料為苯乙烯-二乙烯基苯共聚物。
基本原理:GPC是一種特殊的液相色譜,所用儀器實際上就是一臺高效液相色譜(HPLC)儀,主要配置有輸液泵、進樣器、色譜柱、濃度檢測器和計算機數據處理系統。
與HPLC最明顯的差別在于二者所用色譜柱的種類(性質)不同:HPLC根據被分離物質中各種分子與色譜柱中的填料之間的親和力而得到分離,GPC的分離則是體積排除機理起主要作用。
當被分析的樣品通過輸液泵隨著流動相以恒定的流量進入色譜柱后,體積比凝膠孔穴尺寸大的高分子不能滲透到凝膠孔穴中而受到排斥,只能從凝膠粒間流過,最先流出色譜柱,即其淋出體積(或時間)最小;中等體積的高分子可以滲透到凝膠的一些大孔中而不能進入小孔,比體積大的高分子流出色譜柱的時間稍后、淋出體積稍大;體積比凝膠孔穴尺寸小得多的高分子能全部滲透到凝膠孔穴中,最后流出色譜柱、淋出體積最大。因此,聚合物的淋出體積與高分子的體積即分子量的大小有關,分子量越大,淋出體積越小。分離后的高分子按分子量從大到小被連續的淋洗出色譜柱并進入濃度檢測器。
PEO與PEG的碳鏈骨架相同,但是其合成原料和封端不同,由于原料的性質,使其產物的分子量和結構都有一定的區別。PEO常是指一端為甲基封端,一端為羥基封端的聚環氧乙烷,而PEG一般是兩端都是羥基封端的聚乙二醇。丁苯橡膠在塑煉時分子量分布的變化:在塑煉過程中定時取樣分析,結果如圖,隨時間的增加,高分子組分裂解增加,GPC曲線向低分子量方向移動,經過25min以后,高分子量組分幾乎完全消失。如果塑煉的目的就是消除該組分,那么25min足夠了,通過GPC數據可以幫助工作人員確定塑煉時間。光譜分析是一種根據物質的光譜來鑒別物質及確定它的化學組成,結構或者相對含量的方法。按照分析原理,光譜技術主要分為吸收光譜,發射光譜和散射光譜三種;按照被測位置的形態來分類,光譜技術主要有原子光譜和分子光譜兩種。紅外光譜屬于分子光譜,有紅外發射和紅外吸收光譜兩種,常用的一般為紅外吸收光譜。紅外光譜的原理,在之前的推送中已經詳細介紹過了,此次著重介紹紅外光譜在高分子材料研究中的應用。主要有以下兩種:
1)聚合物的分析與鑒別。聚合物的種類繁雜,譜圖復雜。不同的物質,其結構不一樣,對應的圖譜也不同,因此可以根據分析結果與標準譜圖進行對比才能得到最終結果。
2)聚合物結晶度的測度。由于完全結晶聚合物的樣品很難獲得,因此不能僅用紅外吸收光譜獨立測量結晶度的絕對量,需要聯合其他測試方法的結果。
光照射樣品分子或原子時,外層電子吸收一定波長紫外光,由基態躍遷至激發態而產生的光譜。紫外光波長范圍是10-400 nm。波長在10-200 nm范圍內的稱為遠紫外光,波長在200-400 nm的為近紫外光。對于物質結構表征主要在紫外可見波長范圍,即200-800 nm。1) 定性分析:尤其適用于共軛體系的鑒定,推斷未知物的骨架結構,還可以與紅外光譜、核磁共振波譜法等配合進行定性鑒定和結構分析。比較吸收光譜曲線與最大吸收波長的關系,進行定性測試。2) 定量分析:根據Lambert-Beer定律,一定波長處被測定物質的吸光度與物質的溶度呈線性關系。通過測定溶液對一定波長入射光的吸光度求出該物質在溶液中的濃度和含量。質譜是指廣泛應用于各個學科領域中通過制備、分離、檢測氣相離子來鑒定化合物的一種專門技術。質譜法在一次分析中可提供豐富的結構信息,將分離技術與質譜法相結合是分離科學方法中的一項突破性進展。在眾多的分析測試方法中,質譜學方法被認為是一種同時具備高特異性和高靈敏度且得到了廣泛應用的普適性方法。質譜是提供有機化合物分子量與化學式的方便與可靠方法,也是鑒別有機化合物的重要手段。原理:試樣氣化后,氣體分子進入電離室。電離室的一端安有陰極燈絲,燈絲通電后產生電子束。分子在電子束沖擊下,失去電子,解離成離子和進一步被打碎為不同質量數的帶電荷碎片離子,這樣的離子源是質譜儀最常用的,稱為“電子轟擊離子源”。
1) 高分子材料的單體、中間體以及添加劑的分析。如下圖的質譜圖,可以確定該未知物分子含有1個羧基和1個甲基,其余部分只能是-CO2或者-C3H4。不過后者的可能性更大些。2) 聚合物的表征。每個高分子化合物都具有不同的分子式和分子結構,質譜圖就像是高分子材料的“身份證”。根據其質譜圖,可以確定是哪種高分子材料。X射線是一種波長很短(約為10-8~10-12米),介于紫外線和伽馬射線之間的電磁輻射。由德國物理學家倫琴于1895年發現。X射線能夠穿透一定厚度的物質,并能使熒光物質發光、照相膠乳感光、氣體電離。XRD可應用于高分子結晶度的應用或者計算。天然纖維素結晶度的計算公式有以下四種(劉治剛,中國測試):根據下圖可以看出,天然纖維素的4個衍射晶面半峰寬較大,衍射峰重合度較高,晶相與非晶相重合度較大,導致無定型峰定位困難。
天然纖維素的XRD圖
小角X射線衍射(SAXS)—晶體在原子尺寸上的排列
晶體中的原子在射入晶體的X射線的作用下被迫強制振動,形成一個新的X射線源發射次生X射線。
如果被照射試樣具有不同電子密度的非周期性結構,則次生X射線不會發生干涉現象,該現象被稱為漫射X射線衍射。X射線散射需要在小角度范圍內測定,因此又被稱為小角X射線散射。
①塊狀試樣:塊狀試樣太厚,光束無法通過,因此必須減薄;②薄膜試樣:如薄膜試樣厚度不夠,可以用幾片相同的試樣疊加在一起測試;③粉末試樣:粉末試樣應研磨成無顆粒感,測試時,需用非常薄的鋁箔(載體)包住,或把粉末均勻攪拌在火棉膠中,制成合適厚度的片狀試樣;④纖維試樣:對于纖維狀試樣,應盡可能剪碎,如同粉末試樣那樣制備;⑤顆粒狀試樣:對于無法研磨的粗顆粒狀試樣是比較麻煩的。一個方法是將顆粒盡可能切割成相同厚度的薄片,然后整齊的平鋪在膠帶上;另一個方法是將顆粒熔融或溶解,制成片狀試樣,但前提是不能破壞試樣原有的結構;⑥液體試樣:溶液試樣須注入毛細管中測試。制備溶液時,需注意:溶質在溶劑中完全溶解,即無沉淀;溶質與溶劑的電子密度差盡可能大。在天然的和人工合成的高聚物中,普遍存在小角X射線散射現象,并有許多不同的特征。小角X射線散射在高分子中的應用主要包括以下幾個方面:①通過Guinier散射測定高分子膠中膠粒的形狀、粒度以及粒度分布等;②通過Guinier散射研究結晶高分子中的晶粒、共混高分子中的微區(包括分散相和連續相)、高分子中的空洞和裂紋形狀、尺寸及分布等;③通過長周期的測定研究高分子體系中片晶的取向、厚度、結晶百分數以及非晶層的厚度等;⑤通過Porod-Debye相關函數法研究高分子多相體系的相關長度、界面層厚度和總表面積等;
經過2500℃碳化處理的PAN基碳纖維的試樣的試樣呈現典型的微孔-石墨兩相結構,微孔有明銳界面,石墨基體結構均勻,沒有微密度起伏;經過1340℃碳化處理的PAN基碳纖維的呈現對Porod規律的正偏離,表明碳纖維中除存在微孔外,在亂層石墨基體上存在1 nm以下小尺寸的微密度不均勻區。
導熱系數也叫導熱率,是指在穩定傳熱條件下,1m厚的材料,兩側表面的溫差為1度(K,℃),在1秒鐘內(1s),通過1平方米面積傳遞的熱量,單位為瓦/米·度(W/(m·K),此處為K可用℃代替)。導熱系數是表示材料熱傳導能力大小的物理量。導熱系數是針對均質材料而言的,對于多孔、多層、多結構、各向異性材料,可以叫做平均導熱系數。導熱系數與材料的種類、結構、密度、濕度、溫度、壓力等因素有關。熱擴散系數的定義:熱擴散系數又叫導溫系數,它表示物體在加熱或冷卻中,溫度趨于均勻一致的能力,單位為平方米/秒( m2/s) 。在導熱系數高的物質中熱能擴散的很快,而導熱系數低的物質中熱能則擴散的較慢。這個綜合物性參數對穩態導熱沒有影響,但是在非穩態導熱過程中,它是一個非常重要的參數。熱擴散系數是表示材質均溫能力大小的物理量。熱擴散系數與材料的導熱系數、密度、比熱容等因數有關。熱擴散系數采用非穩態(瞬態)法測量,在穩態導熱中不起影響。激光閃射法原理:激光法直接測試的是材料的熱擴散系數,其基本原理示意圖如下:在爐體控制的一定溫度下,由激光源發射光脈沖均勻照射在樣品下表面,使試樣均勻加熱,通過紅外檢測器鏈接測量樣品上表面相應溫升過程,得到溫度(檢測器信號)升高和時間的關系曲線。瞬態法適用于高導熱系數的材料,如金屬、合金、陶瓷以及多層材料等。
穩態熱流法原理:將一定厚度的樣品放入兩個平板間,在其垂直方向通入一個恒定的單向熱流,使用校正過的熱流傳感器測量通過樣品的熱流,傳感器在平板與樣品之間與樣品接觸。當冷板和熱板的溫度穩定后,測得樣品厚度、樣品上下表面的溫度和通過樣品的熱流量,根據傅里葉定律即可確定樣品的導熱系數。該法適用于導熱系數較小的固體材料、纖維材料與多空隙材料,例如各種保溫材料。
導熱系數測試方法測試標準:針對材料導熱系數測試,除了需要有對應的測試方法和測試設備,還需要有對用的標準來規范測試方法、測試過程、測試條件、測試樣品、測試范圍等信息。在材料導熱系數的測試領域,常用的導熱系數測試標準主要采用美國材料試驗協會(ASTM)的 ASTM-D5470,ASTM-E1461,ASTM-E1530,ASTM C518-04等。不同導熱材料特點:對于電子器件而言,高分子絕緣材料具有獨特的結構和易改性、易加工的特點,使其具有其他材料不可比擬、不可取代的優異性能。但是一般高分子材料都是熱的不良導體,其導熱系數一般都低于 0.5 Wm-1K-1。一些常見的高分子在室溫下的導熱系數如下表所示。
基本概念:在拉伸試驗中,保持這種受力狀態最終,就是測量拉伸力直至材料斷裂為止,所承受的最大拉伸應力稱為拉伸強度。
實驗原理:拉伸實驗是對材料沿縱軸方向施加靜態拉伸負荷,使其破壞。通過測定試樣的屈服力破壞力和材料標距間的伸長來求得材料的屈服強度、拉伸強度和伸長率。
基本定義:
拉伸應力:試樣在計量標距范圍內,單位初始橫截面上承受的拉伸負荷。拉伸強度:在拉伸試驗中試樣直到斷裂為止,所承受的最大拉伸應力。拉伸斷裂應力:在拉伸應力-應變曲線上,斷裂時的應力。拉伸屈服應力:在拉伸應力-應變曲線上,屈服點處的應力。斷裂伸長率:在拉力作用下,試樣斷裂時,標線間距離的增加量與初始標距之比,以百分率表示。
彈性模量:在比例極限內,材料所受應力與產生響應的應變之比。
由應力-應變的相應值彼此對應的繪成曲線,通常以應力值作為縱坐標,應變值作為橫坐標。應力-應變曲線一般分為兩個部分:彈性變形區和塑性變形區,在彈性變形區,材料發生可完全恢復的彈性變形,應力和應變呈正比例關系。曲線中直線部分的斜率即是拉伸彈性模量值,它代表材料的剛性。彈性模量越大,剛性越好。在塑性變形區,應力和應變增加不在呈正比關系,最后出現斷裂。
③測量試樣中間平行部分的厚度和寬度,精確0。01mm,II型試樣中間平行部分的寬度,精確到0。05mm,測3點,取算術平均值。④夾具夾持試樣時,要使試樣縱軸與上下夾具中心連線重合,且松緊適宜。⑥記錄屈服時負荷,或斷裂負荷及標距間伸長。試樣斷裂在中間平行部分之外時,此試樣作廢,另取試樣補做。
(1)成型條件:由試樣自身的微觀缺陷和微觀不同性引起(3)拉伸速度:塑料屬于粘彈性材料,其應力松弛過程與變形速率緊密相關,需要一個時間過程(4)預處理:材料在加工過程中,由于加熱和冷卻的時間和速度不同,易產生局部應力集中,經過在一定溫度下的熱處理或稱退火處理,可以消除內應力,提高強度(5)材料性質:結晶度、取向、分子量及其分布、交聯度
基本概念:試樣破壞時的最大壓縮載荷除以試樣的橫截面積,稱為壓縮強度極限或抗壓強度。壓縮試驗是測定材料在軸向靜壓力作用下的力學性能的試驗,是材料機械性能試驗的基本方法之一。與拉伸試驗相似,通過壓縮試驗可以作出壓縮曲線。
壓縮實驗室最常見的一種力學試驗,是鈀試樣置于萬能試驗機的兩壓板之間,并在沿試樣兩端面的主軸方向,以恒定速率施加一個可以測量的大小相等相反的力,并使試樣沿軸向方向縮短,而徑向方向增大,產生壓縮變形,直到試樣破裂或者變形達到規定的如25%的數值為止。施加的負荷由試驗機上直接讀得,并按下式計算其壓縮應力。式中:σ-壓縮應力,MPa;P-壓縮負荷,N;F-試樣原始橫截面積,mm2。試樣在壓縮負荷作用下高度的該變量稱為壓縮變形,按下式計算:式中:ΔH-試樣的壓縮形變,mm;H0-試樣原始高度,mm;H-壓縮過程中任何時刻試樣的高度,mm。式中:ε-試樣壓縮應變;ΔH-試樣的壓縮形變,mm;H0-試樣原始高度,mm;H-壓縮過程中任何時刻試樣的高度,mm。
壓縮屈服應力:指應力-應變曲線上第一次出現應變增加而應力不增加的轉折點(屈服點)對應的應力,以MPa表示。
壓縮強度:指在壓縮試驗中試樣承受的最大壓縮應力,以MPa表示,它不一定市試樣破壞瞬間所承受的壓縮應力。
定應變壓縮應力:指規定應變時的壓縮應力,即與應變為25%時對應的應力值,以MPa表示。
壓縮模量:指在應力-應變曲線的線性范圍內,壓縮應力與壓縮應變的比值,以MPa表示,取應力-應變曲線上兩點的應力差與對應的應變之比。
(1)試樣的細長比:(試樣高度與試樣截面的最小回轉半徑之比)是最大的影響因素。由于試樣受壓時,其上下端面與試驗機壓板之間產生較大的摩擦力,阻礙試樣上下兩端面的橫向變形,試樣高度越小,影響程度越大。
(2)實驗速度:一般來講,隨著實驗速度的增加,壓縮強度與壓縮應變值均有所增加。實驗速度在1-5mm/min時變化較小。壓縮試驗的同一試樣必須在同一實驗速度下進行,并且最好選用較低的實驗速度。
試驗時將一規定形狀和尺寸的試樣置于兩支坐上,并在兩支坐的中點施加一集中負荷,使試樣產生彎曲應力和變形。這種方法稱靜態三點式彎曲試驗。(另一加載方法為四點式,這里不介紹。)
撓度:彎曲試驗過程中,試樣跨度中心的定面或底面偏離原始位置的距離。彎曲應力:試樣在彎曲過程中的任意時刻,中部截面上外層纖維的最大正應力。彎曲強度:在到達規定撓度值時或之前,負荷達到最大值時的彎曲應力。定撓彎曲應力:撓度等于試樣厚度1.5倍時的彎曲應力。彎曲屈服強度:在負荷-撓度曲線上,負荷不增加而撓度驟增點的應力。
①使用游標卡尺測量試樣中間部位的寬度和厚度,測量三點,取其平均值,精確到0.02mm。③調節好跨度,將試樣放于支架上,上壓頭與試樣寬度的接觸線須垂直于試樣長度方向,試樣兩端緊靠支架兩頭。④啟動下降按鈕,試驗機按設定的參數開始工作。當壓頭接觸到試樣后,計算機開始自動記錄試樣所受的載荷及其產生的位移數據。至試樣到達屈服點或斷裂時為止,立即停機。⑤保存數據,并根據數據作彎曲載荷-位移曲線圖,并保存。根據圖形分析試樣的彎曲力學行為。
(1)試樣尺寸和加工試樣的厚度和寬度都與彎曲強度和撓度有關。(2)加載壓頭半徑和支座表面半徑如果加載壓頭半徑很小,對試樣容易引起較大的剪切力而影響彎曲強度。支座表面半徑會影響試樣跨度的準確性。(3)應變速率彎曲強度與應變速率有關,應變速率較低時,其彎曲強度也偏低。(4)試驗跨度當跨厚比增大時,各種材料均顯示剪切力的降低,可見用增大跨厚比可減少剪切應力,使三點彎曲試驗更接近純彎曲。(5)溫度就同一種材料來說,屈服強度受溫度的影響比脆性 強度的大。現行塑料彎曲性能實驗的國家標準為GB/T9341-2008。
沖擊試驗是用來度量材料在高速沖擊狀態下的韌性或對斷裂的抵抗能力。
一般沖擊實驗采用三種方法:
(1)擺錘式:試驗安放形式有簡支梁式(charpy)----支撐試樣兩端而沖擊中部;懸臂梁式(Izod)---試樣一端固定而沖擊自由端。
(3)高速拉伸法。此方法雖較理想,可直接轉換成應力—應變曲線,計算曲線下的面積,便可得沖擊強度,還可定性判斷是脆性斷裂還是韌性斷裂,但對拉力機要求較高。
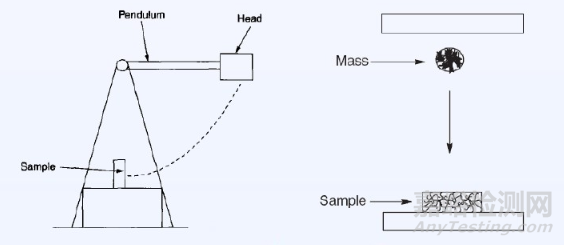
④擺錘和支架軸、擺錘刀口和試樣相互摩擦損失的能量。
⑤擺錘運動時,試驗機固有的能量損失。
(2)溫度和適度 材料的沖擊性能測試依賴于溫度。
(3)試樣尺寸 使用同一配方和同一成型條件而厚度不同的材料做沖擊試驗時,所得的沖擊強度不同。
(4)沖擊速度 擺錘的沖擊速度高時沖擊強度的數值反而降低。
高分子材料具有大分子鏈結構和特有的熱運動,決定了它具有與低分子材料不同的物理性態。高分子材料的力學行為最大特點是它具有高彈性和粘彈性。在外力和能量作用下,比金屬材料更為強烈地受到溫度和時間等因素的影響,其力學性能變化幅度較大。
高聚物受力產生的變形是通過調整內部分子構象實現的。由于分子鏈構象的改變需要時間,因而受力后除普彈性變形外,高聚物的變形強烈地與時間相關,表現為應變落后于應力。除瞬間的普彈性變形外,高聚物還有慢性的粘性流變,通常稱之為粘彈性。高聚物的粘彈性又可分為靜態粘彈性和動態粘彈性兩類。
靜態粘彈性指蠕變和松弛現象。與大多數金屬材料不同,高聚物在室溫下已有明顯的蠕變和松弛現象。
高分子材料的蠕變即在一定溫度和較小的恒定外力(拉力、壓力或扭力等)作用下、高分子材料的形變隨時間的增加而逐漸增大的現象。
蠕變過程及原理:下圖蠕變曲線,t1是加荷時間,t2是釋荷時間。從分子運動和變化的角度來看,蠕變過程包括下面三種形變:當高分子材料受到外力作用時,分子鏈內部鍵長和鍵角立刻發生變化,這種形變量是很小的,稱為普彈形變。當分子鏈通過鏈段運動逐漸伸展發生的形變,稱為高彈形變。如果分子間沒有化學交聯,線形高分子間會發生相對滑移,稱為粘性流動。這種流動與材料的本體粘度有關。在玻璃化溫度以下鏈段運動的松弛時間很長,分子之間的內摩擦阻力很大,主要發生普彈形變。在玻璃化溫度以上,主要發生普彈形變和高彈形變。當溫度升高到材料的粘流溫度以上,這三種形變都比較顯著。由于粘性流動是不能回復的,因此對于線形高聚物來說,當外力除去后會留下一部分不能回復的形變,稱為永久形變。
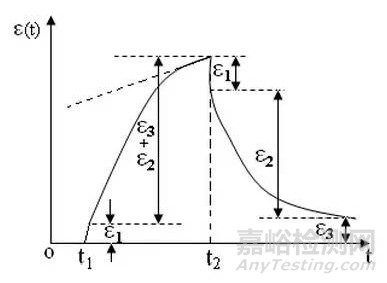
蠕變與溫度高低和外力大小有關,溫度過低,外力太小,蠕變很小而且很慢,在短時間內不易覺察;溫度過高、外力過大,形變發展過快,也感覺不出蠕變現象;在適當的外力作用下,通常在高聚物的玻璃化溫度以上不遠,鏈段在外力下可以運動,但運動時受到的內摩擦力又較大,只能緩慢運動,則可觀察到較明顯的蠕變現象。
蠕變是材料彈性與粘性的相互作用結果,材料彈性好,其蠕變應力大,蠕變溫度高,如果粘性大于彈性則反之,讓我們可以把握實際可能出現的情況。所以一般來講,特別是塑料或者壓敏膠,材料加工要蠕變好(流動性好),而材料應用則需蠕變差(材料穩定,蠕變意味著材料發生變化)。蠕變涉及到材料結構、分子量、分子鏈等等因素。避免蠕變,就是想方法讓材料穩定,如提高彈性讓臨界應力大,添加抗氧劑讓耐老化性能好,分子結構穩定。
應力松弛:就是在固定的溫度和形變下,聚合物內部的應力隨時間增加而逐漸衰減的現象。這種現象也在日常生活中能觀察到,例如橡膠松緊帶開始使用時感覺比較緊,用過一段時間后越來越松。也就是說,實現同樣的形變量,所需的力越來越少。未交聯的橡膠應力松弛較快,而且應力能完全松弛到零,但交聯的橡膠,不能完全松弛到零。
線形聚合物的應力松弛的分子機理如圖所示,拉伸時張力迅速作用使纏繞的分子鏈伸長,但這種伸直的構象時不平衡的,由于熱運動分子鏈會重新卷曲,但形變量被固定不變,于是鏈可能解纏結而轉入新的無規卷曲的平衡態,于是應力松弛為零。交聯聚合物不能解纏結,因而應力不能松弛到零。
目的:測量材料的適用性,間接了解材料的磨擦性能、拉伸性能、固化程度等力學性能
常用的硬度測試方法:邵氏硬度、洛氏硬度,硬度體現的是產品的堅硬程度。在施加荷重的狀態下,測定堅硬的圓珠凹陷時的抗衡性的實驗。如果塑料中膠含量較多的話,沖擊強度將會增加,但硬度會下降。
布氏硬度起源于1900年,是最早被廣泛接受的測試方法。它的原理是使用球壓頭D通過一定的壓力F在材料表面產生壓痕,再用光學系統測量壓痕直徑d,最后通過經驗公式計算可得到布氏硬度值,如圖所示。

④對樣品表面施加力F(必須緩慢增加,且適當保持以保證塑性變形)
計算公式如下:
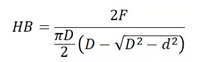
布氏硬度早期的壓頭材料為硬化鋼(符號HBS),不適用于非常硬(>450HB)的材料,容易變形,最終會導致測試結果誤差大。ISO和ASTM標準先后推薦使用WC(碳化鎢,符號HBW)球來做所有的布氏硬度測試適用測試范圍達650HB,更加耐用。壓痕過小,壓痕難以準確的測量,并且壓頭損壞的風險變大;壓痕過大,硬度測試的靈敏度降低。因此,標準規定了壓痕尺寸的上下限,壓痕直徑與壓頭直徑的比值d/D應在0.24和0.6之間。在測試過程中要注重選擇合適的球壓頭直徑和載荷大小與被測試的材料匹配。硬度測試產生壓痕的過程會引起殘余應力和加工硬化,過于靠近邊緣會導致材料不足以限制壓痕周圍的變形區,因此,若壓痕位置不當,測試結果易產生能夠誤差。
布氏硬度測試要選擇好標尺(載荷大小與球壓頭直徑的組合)。通常采用壓痕相似性原理來設計布氏硬度的標尺。
壓痕相似性原理:對于不同的壓頭直徑和載荷組合,當壓入角相同時,材料的硬度值才具有可比性。對于相同的材料,當載荷與壓頭直徑平方的比值(K=F/D² )相同時,壓入角相同,所求得的硬度值也相同。
壓入角28°與74°之間硬度值變化很小,44°(d/D=0.375)是最理性的狀況。對同一材料而言160HBW10/500大約相當于180HBW10/3000。因此,常見的比值,不同比值下測量結果不具備可比性。如表2,HB30與HB10、HB5標尺下所得的硬度值不可以做比較。
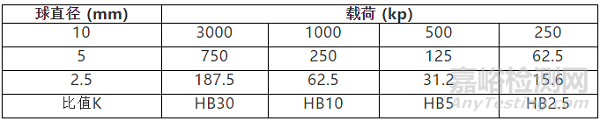
K值得選擇具有一定的要求,通常高硬度材料選用較大K值,低硬度材料選用較小K值。常用的K值選擇如表3。
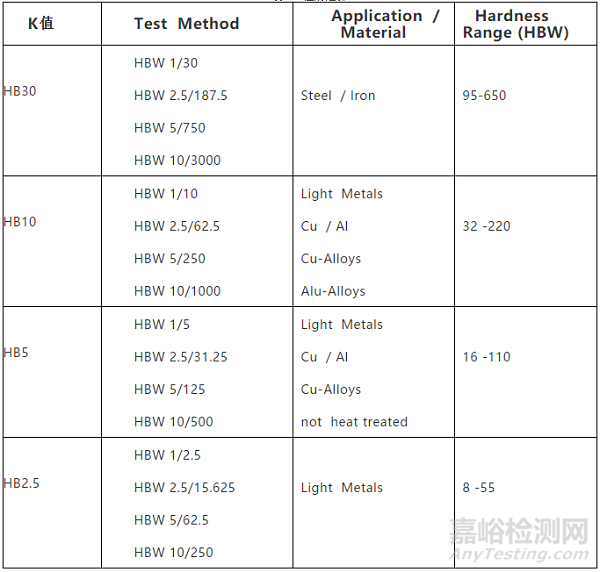
布氏硬度測試過程中,試驗力易引起樣品的扭曲、變形和移動,導致誤差的產生。因此ASTM樣品厚度要達到壓痕深度的10倍。不同標尺要求的樣品最小厚度如表4所示。
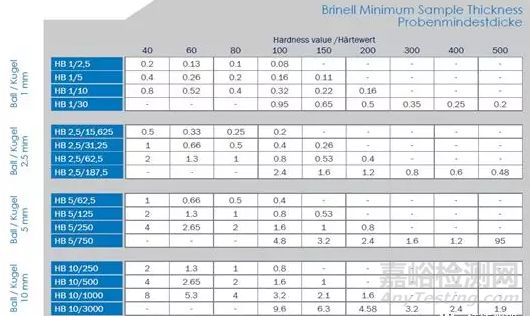
相對較寬的測試范圍8~650HBW;不同測試力之間硬度值可以對比 (對于相同的F/D2)壓痕較大;對樣品表面狀況不敏感;測試大塊材料硬度;對光學測量誤差不敏感;非常適合于大晶粒材料的測試;與拉伸強度之間有較好的對應關系。大的壓痕意味著不能區分出局部區域的硬度值變化;不適合于涂層和薄片樣品的測試;不適合于高硬度材料的硬度測試;通過光學顯微鏡測量壓痕,不同操作者對測量的影響較大;不建議測試弧形表面。適用于鑄鐵、鍛造件、各種退火及調質的鋼材,有色金屬、硬質合金、陶瓷、不銹鋼等,廣泛應用在汽車和航空等行業,試驗室和車間的樣品測試和質量控制。可測平面和圓柱形工件,不宜測定太硬、太小、太薄和表面不允許有較大壓痕的試樣或工件。洛氏硬度起源于20世紀初,是由洛克威爾(S.P.Rockwell)提出來的學術概念。洛氏硬度測試的原理是以壓痕塑性變形深度來確定硬度值。

根據材料選擇一定的標尺(HRA、HRB、HRC等)采用洛氏硬度測試方法時,需要根據不同的標尺和樣品情況來對試樣表面進行處理。
與洛氏硬度相同,壓痕距離邊緣至少為壓痕直徑的2.5倍(并且ISO6508規定距離邊緣不小于1mm),兩壓痕之間的距離至少為壓痕直徑的3倍。洛氏硬度測試對樣品尺寸有要求,樣品厚度不能小于殘余壓痕深度的10倍,且背面不能出現明顯的變形痕跡。由此樣品的厚度決定了載荷的選擇,載荷必須保證其所引起的變形小于樣品的最小厚度。對于每一種硬度試驗,都存在最小可測量厚度。根據不同壓頭類型,分別為:
使用金剛石壓頭時,樣品最小厚度為壓痕深度的10倍
壓頭尖端破裂(通常導致硬度讀數偏高);被測試材料或者夾具軌道的變形(通常導致硬度讀數偏低);過大的振動(通常導致硬度讀數偏低);表面制備質量差(注意制備也包括測試零件的底部和試臺);壓頭和樣品表面的壓入角度(不能超過2o 偏差)不需要通過光學測量就可以得到硬度值;快速簡單;不同操作者對結果影響不大;表面粗糙度對結果影響較小。洛氏硬度測試具有30種不同的標尺,幾乎覆蓋所有材料的硬度測試,以下是其一些典型的應用:常規洛氏:端淬試驗、齒輪、鋼棒、緊固件、汽車部件、軸承、熱處理工件、硬質合金等。
表面洛氏:薄鋼板、線、小直徑棒、鍍錫鋼片、涂層、硬化層。
通過SEM對材料斷口、裂紋、磨痕等進行觀察,進而評價其力學性能
自從1965年第一臺商品掃描電鏡問世以來,經過40多年的不斷改進,掃描電鏡的分辨率從第一臺的25nm提高到現在的0.01nm,而且大多數掃描電鏡都能與X射線波譜儀、X射線能譜儀等組合,成為一種對表面微觀世界能夠經行全面分析的多功能電子顯微儀器。在材料領域中,掃描電鏡技術發揮著極其重要的作用,被廣泛應用于各種材料的形態結構、界面狀況、損傷機制及材料性能預測等方面的研究。
掃描電鏡由電子槍發射出來的電子束,在加速電壓的作用下,經過磁透鏡系統匯聚,形成直徑為5nm,經過二至三個電磁透鏡所組成的電子光學系統,電子束會聚成一個細的電子束聚焦在樣品表面。在末級透鏡上邊裝有掃描線圈,在它的作用下使電子束在樣品表面掃描。由于高能電子束與樣品物質的交互作用,結果產生了各種信息:二次電子、背反射電子、吸收電子、X射線、俄歇電子、陰極發光和透射電子等。這些信號被相應的接收器接收,經放大后送到顯像管的柵極上,調制顯像管的亮度。由于經過掃描線圈上的電流是與顯像管相應的亮度一一對應,也就是說,電子束打到樣品上一點時,在顯像管熒光屏上就出現一個亮點。
掃描電鏡就是這樣采用逐點成像的方法,把樣品表面不同的特征,按順序,成比例地轉換為視頻信號,完成一幀圖像,從而使我們在熒光屏上觀察到樣品表面的各種特征圖像。
波譜儀是利用布拉格方程2dsinθ=λ,從試樣激發出了X射線經適當的晶體分光,波長不同的特征X射線將有不同的衍射角2θ。波譜儀是微區成分分析的有力工具。波譜儀的波長分辨率是很高的,但是由于X射線的利用率很低,所以它使用范圍有限。能譜儀是利用X光量子的能量不同來進行元素分析的方法,對于某一種元素的X光量子從主量子數為n1的層躍遷到主量子數為n2的層上時,有特定的能量ΔE=En1-En2。能譜儀的分辨率高,分析速度快,但分辨本領差,經常有譜線重疊現象,而且對于低含量的元素分析準確度很差。 (2) 能譜儀的結構比波譜儀簡單,沒有機械傳動部分,因此穩定性和重復性都很好。
(3) 能譜儀不必聚焦,因此對樣品表面沒有特殊要求。
但是能譜儀的分辨率比波譜儀低;能譜儀的探頭必須保持在低溫狀態,因此必須時時用液氮冷卻。
1) 觀察材料的界面形態:高分子的表面物理形態和化學結構是決定材料性能的基本因素,也是影響高分子材料的摩擦性能、光學性能、吸水性和生物相容性等的主要因素。
2) 聚合物的增韌機理研究中的應用:高分子聚合物的斷裂一般分為脆性斷裂和韌性斷裂。脆性斷裂的斷面較光滑而韌性斷裂的斷面較粗糙,聚合物的增韌就是把聚合物的斷裂方式由脆性斷裂轉變為韌性斷裂,使聚合物在受到拉伸時有較高的斷裂伸長率,在受到沖擊時不易破壞。高分子材料脆性斷面表面的斷裂源,鏡面區(1)、霧狀區(2)和粗糙區(3)
TEM——基本形貌、結晶結構、高分子的組裝形貌等信息。
在光學顯微鏡下無法看清小于0.2微米的細微結構,這些結構稱為亞顯微結構或超細結構。要想看清這些結構,就必須選擇波長更短的光源,以提高顯微鏡的分辨率。
電子顯微鏡(Transmission electronmicroscope,縮寫TEM),簡稱透射電鏡,是把經加速和聚集的電子束投射到非常薄的樣品上,電子與樣品中的原子碰撞而改變方向,從而產生立體角散射。散射角的大小與樣品的密度、厚度相關,因此可以形成明暗不同的影像。通常,透射電子顯微鏡的分辨率為0.1~0.2nm,放大倍數為幾萬~百萬倍,用于觀察超微結構,即小于0.2微米、光學顯微鏡下無法看清的結構,又稱“亞顯微結構”。
來源:《Characterization Techniquesof Nanomaterials》[書]電子槍:發射電子。由陰極,柵極和陽極組成。陰極管發射的電子通過柵極上的小孔形成射線束,經陽極電壓加速后射向聚光鏡,起到對電子束加速和加壓的作用。中間鏡:二次放大,并控制成像模式(圖像模式或者電子衍射模式)。
CCD相機:電荷耦合元件,將光學影像轉化為數字信號。
在實際操作TEM之前,要求所測試樣必須滿足一定的條件,針對不同類型的試樣有不同的制取方法。
高分子材料的特殊制樣方法
聚焦離子束技術(Focused Ion beam,FIB)是近年來發展起來的新技術,它是利用電透鏡將離子束聚焦成非常小尺寸的離子束轟擊材料表面,實現材料的剝離、沉積、注入、切割和改性。聚焦離子束技術(FIB)利用高強度聚焦離子束對材料進行納米加工,配合高倍數電子顯微鏡實時觀察,成為了納米級分析、制造的主要方法。要保證樣品的純度,不受環境和人為制樣處理的污染,一般使用FIB進行樣品的制備。FIB是一種專業的制樣方法,與人工制樣的人為影響因素多等缺點相比,FIB能夠觀察到樣品缺陷與基材的界面情況,利用FIB就可以準確定位切割,制備缺陷位置截面樣品,完全滿足對制樣的需求。聚焦離子束(FIB)技術是利用高強度聚焦離子束對材料進行納米加工,實現樣品表面分子和元素種類的空間分布信息,成為了納米級分析、制造的主要方法。(3)以無機成分為主,否則會造成電鏡嚴重的污染,高壓跳掉,甚至擊壞高壓槍;(1)需要電解減薄或離子減薄,獲得幾十納米的薄區才能觀察;(2)如晶粒尺寸小于1μm,也可用破碎等機械方法制成粉末來觀察;(4)塊狀樣品制備復雜、耗時長、工序多、需要由經驗的老師指導或制備;樣品的制備好壞直接影響到后面電鏡的觀察和分析。所以塊狀樣品制備之前,最好與TEM的老師進行溝通和請教,或交由老師制備。理論:根據電子顯微學理論,加速電壓越高,理論空間分辨率越高。 缺陷:對于不同的試樣,高加速電壓同時會帶來輻照損傷等問題,影響實際分辨率。
影響因素:加速電壓固定后,影響透射電子顯微鏡分辨率的因素可歸結為球差、象散和色差。
偏光顯微鏡是研究晶體光學性質的重要儀器,同時又是其他晶體光學研究法(油浸法、弗氏臺法等)的基礎。偏光顯微鏡是利用光的偏振特性對具有雙折射性物質進行研究鑒定的必備儀器,可做單偏光觀察,正交偏光觀察,錐光觀察。將普通光改變為偏振光進行鏡檢的方法,以鑒別某一物質是單折射(各向同行)或雙折射性(各向異性)。雙折射性是晶體的基本特征。因此,偏光顯微鏡被廣泛地應用在礦物、化學等領域。

研究晶體光學性質所使用的顯微鏡裝有起偏鏡(下偏光鏡、前偏光鏡)和檢偏鏡(上偏光鏡、后偏振鏡、分析鏡)。自然光經起偏鏡后成為在某一固定方向上振動的偏振光。由于裝有起偏鏡和檢偏鏡,故將此類顯微鏡稱為偏光顯微鏡。
光線從偏振片通過呈光學各向同性的聚合物熔融態或無定形態時不改變偏振方向,因此,用偏光顯微鏡觀察時,視野是全暗的。光線通過呈光學各向異性的聚合物結晶態或取向態時會分解成偏振方向相互垂直的兩束光。因此,用偏振顯微鏡觀察時,會呈現特征的視野。
偏光顯微鏡應用實例之球晶觀察:
當α=0°、90°、180°和270°時,sin2α為0,這幾個角度沒有光線通過;當α為45°的奇數倍時,sin2α有極大值,因而視野最亮。于是,球晶在正交偏光顯微鏡下呈現特有的消光十字圖像。
Ziegler-Natta催化劑合成的等規聚丙烯等溫結晶形成的球晶
在一定溫度下,球晶的生長是等速的,用偏光顯微鏡可以進行等溫結晶動力學的研究,方法是測定球晶半徑隨時間變化的關系。

掃描探針顯微鏡(Scanning Probe Microscope,SPM)是掃描隧道顯微鏡及在掃描隧道顯微鏡的基礎上發展起來的各種新型探針顯微鏡(原子力顯微鏡,靜電力顯微鏡,磁力顯微鏡,掃描離子電導顯微鏡,掃描電化學顯微鏡等)的統稱,是國際上近年發展起來的表面分析儀器,是綜合運用光電子技術、激光技術、微弱信號檢測技術、精密機械設計和加工、自動控制技術、數字信號處理技術、應用光學技術、計算機高速采集和控制及高分辨圖形處理技術等現代科技成果的光、機、電一體化的高科技產品。
掃描探針顯微鏡原理:基于量子的隧道效應,利用探針與樣品在近距離(<0.1 nm)時,由于二者存在電位差而產生隧道電流,隧道電流對距離非常敏感,控制探針在被檢測樣品的表面進行掃描,同時記錄下掃描過程中探針尖端和樣品表面的相互作用,就能得到樣品表面的相關信息。顯然,利用這種方法得到被測樣品表面信息的分辨率取決于控制掃描的定位精度和探針作用尖端的大小(即探針的尖銳度)。
STM要求掃描的范圍從10 nm到1微米以上,可以用來觀察原子水平的樣品形貌。
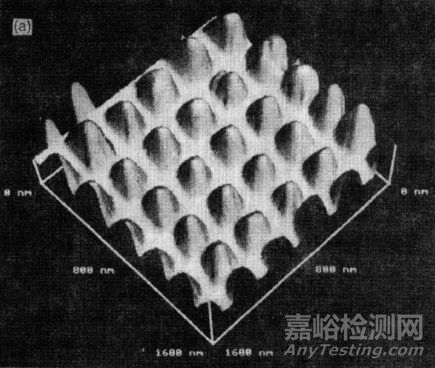
有嚴重缺陷和較為完美的高分子鍍膜(單位:nm)
掃描探針顯微鏡的應用:掃描探針顯微鏡正在迅速地被應用于科學研究的許多領域,如納米技術,催化新材料,生命科學,半導體科學等。如材料表面形貌、相組成分析;材料表面各種缺陷、污染情況分析;材料表面力性能研究;材料表面電、磁性能研究。
材料科學是科學技術發展的重要基礎學科之一, 其中, 高分子材料作為材料科學中一個重要的研究方向, 已經滲入到生活以及工業領域內的方方面面, 發揮著不可替代的重要作用。科學技術的飛速發展, 對高分子材料的需求也日益增長, 同時, 也對高分子材料的性能提出了更高的要求, 高性能化以及復合化成為了高分子材料發展的重要方向。




















































