您當前的位置:檢測資訊 > 科研開發(fā)
嘉峪檢測網(wǎng) 2024-06-23 16:06
1、不同藥典中不溶性微粒檢測的歷史發(fā)展
1.1 中國藥典
1985年版《中華人民共和國藥典》收錄了100 mL以上大容量注射液的不溶性微粒檢測(顯微計數(shù)法);2000年版《中華人民共和國藥典》增加了光阻法作為不溶性微粒檢測的第二法;2005年版《中華人民共和國藥典》將光阻法由第二法改成第一法,并增加100 mL以下小容量注射液劑型的檢測方法;2010年版《中華人民共和國藥典》通則<0903>更新,光阻法和顯微計數(shù)法的檢測限度標準與美、日、歐藥典基本一致;2015年版《中華人民共和國藥典》增加了用于無菌粉末、無菌原料藥、注射用濃溶液的檢測方法;2020年版《中華人民共和國藥典》通則<0903>內(nèi)容與2015年版內(nèi)容保持一致。
1.2 美國藥典
1975年美國藥典(USP)采用顯微計數(shù)法檢測評估100 mL以上大容量注射劑中不溶性微粒;1985年增加光阻法為第二法并對100 mL及以下的小容量注射液劑型檢測方法作了要求;1995年USP 23<788>將光阻法改為第一法,顯微計數(shù)法作為第二法,并對判定標準進行了修改且沿用至今;2017年USP 40<1788>收錄不溶性微粒檢測第三法——動態(tài)(流動)圖像法(Flow Imaging Method,FI),但FI作為新興技術(shù),缺乏歷史數(shù)據(jù),難以建立統(tǒng)一的規(guī)范,至今仍未被收錄至通則<788> 中。
1.3 歐洲藥典和日本藥典
對比歐洲藥典5.0版(EP 5.0)和歐洲藥典10.0版(EP 10.0)及日本藥典15版(JP 15)和日本藥典18版(JP 18),發(fā)現(xiàn)歐洲藥典和日本藥典不溶性微粒檢測方法相關(guān)章節(jié)在近年來無明顯變化。但是EP 11.0<2.9.19>章節(jié)中內(nèi)容相較于EP 10.0增加了生物技術(shù)藥物不溶性微粒檢測相關(guān)內(nèi)容。
2、不同藥典中不溶性微粒要求比較
自2010年起,《中華人民共和國藥典》2010年版對不溶性微粒的要求與美國藥典(USP 32)、日本藥典(JP 15)、歐洲藥典(EP 6.0)已基本保持一致,但在具體檢測方法上仍存在部分差異。此外,美國藥典中不溶性微粒檢測除了注射劑通用的USP<788>章節(jié)外,還專門針對治療性蛋白注射劑(USP<787>)和眼用制劑(USP<789>)單設(shè)章節(jié),要求更加清晰。JP 18也針對治療性蛋白注射液單設(shè)章節(jié)<6.17>,而在《中華人民共和國藥典》2020年版中卻未見區(qū)分。以下將從ChP 2020、USP-NF 2023、EP 11.0、JP 18中注射劑不溶性微粒要求的通用章節(jié)的檢測方法、質(zhì)量標準、系統(tǒng)適用性要求三個方面進行比較論述。
2.1 檢測方法的對比
不同藥典推薦的不溶性微粒檢測方法均為光阻法與顯微計數(shù)法,但在檢測涉及的試驗環(huán)境、檢查用溶劑和具體的樣品檢測要求上,中國藥典與美、歐、日藥典存在差異。
2.1.1 對試驗環(huán)境和檢查用溶劑的要求
為了盡可能減小外來顆粒對樣品檢測結(jié)果的影響,各國藥典除了對測試環(huán)境和檢查用溶劑提出要求外,還要求在檢查前進行檢測。USP-NF 2023明確了該檢測的目的是確認測試環(huán)境、玻璃器皿、微粒檢測用水等是否滿足試驗要求,且該檢測可重復(fù)進行直至結(jié)果滿足要求后才可進行后續(xù)試驗。對比來看,美國藥典對于檢查用注射用水的要求較高,具體見表1。

2.1.2 樣品檢測要求
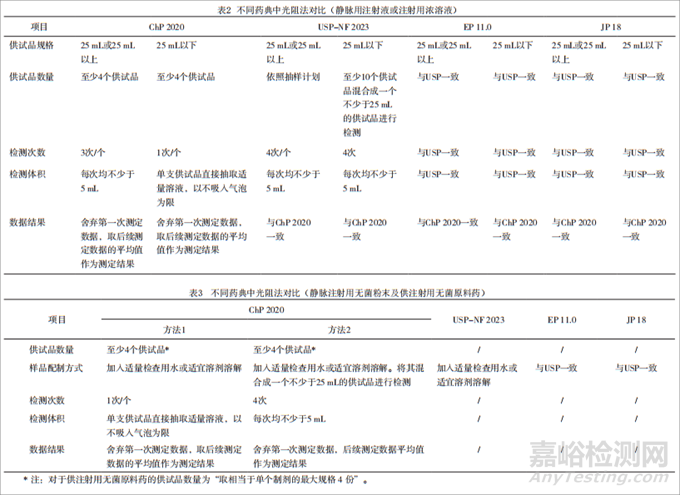
在具體的樣品檢測要求方面,不同藥典在相同供試品規(guī)格下的供試品數(shù)量、檢測次數(shù)、檢測體積有所不同。此外,USP-NF 2023<788>要求對于大容量注射劑,應(yīng)對單個用藥單元進行測試,而這一點在Chp 2020通則<0903>中并未明確。且在FDA官網(wǎng)的GMP問答中,也表示不接受測試單元之間的結(jié)果進行平均,因為顆粒物質(zhì)在整個批次中可能是不均勻地分散的,評估每個單獨單元的結(jié)果的目的是確保代表性并檢測批次內(nèi)的潛在變化。
美、歐、日藥典就不溶性微粒檢測相關(guān)內(nèi)容已達成一致,不會單方面作出修改。通過與中國藥典內(nèi)容對比可見,無論是光阻法還是顯微計數(shù)法,在對于不同規(guī)格的供試品,其供試品數(shù)量和檢測次數(shù)要求上,美、歐、日藥典要求與中國藥典差異較大,尤其是在對25 mL以下的供試品檢測方面。美、歐、日藥典要求更側(cè)重在對25 mL以下的供試品檢測要求的限定,其檢測要求也更為詳細和嚴苛。兩種方式各有優(yōu)劣,比如采用美、歐、日藥典的檢測方法時,供試品數(shù)量要求多,測試覆蓋的樣品體積大,檢測得到的數(shù)據(jù)也更具批代表性;而且,光阻法檢測時,對于儀器是否具備微量進樣功能的依賴度更低。但是,對于單瓶貨值比較高的小容量規(guī)格的供試品而言,測試時供試品用量大,檢測成本較高,而且雖整體樣品量大而更具代表性,但也在一定程度上平均了各個供試品的品質(zhì)差異,弱化了在實際應(yīng)用過程中單瓶供試品間的差異。采用中國藥典的檢測方法,則可以降低企業(yè)的檢測成本,并更有利于監(jiān)測單個供試品內(nèi)不溶性微粒水平的波動。
此外,Chp 2020還明確了靜脈注射用無菌粉末和供注射用無菌原料藥的測試方法,而USP-NF2023中僅提到了粉末的溶解方法。光阻法檢查方法對比見表2、表3,顯微計數(shù)法檢查方法見表4、表5。


2.2 質(zhì)量標準的對比
2.2.1 方法優(yōu)先級比較
不同藥典均將光阻法作為檢測方法的第一法,顯微計數(shù)法作為第二法。美國藥典、日本藥典、歐洲藥典均明確說明光阻法優(yōu)先,中國藥典并未明確提及,且在描述不適合光阻法檢測的特殊樣品時,表述中出現(xiàn)了顯微計數(shù)法測定結(jié)果有效性略優(yōu)于光阻法的描述:“當光阻法測定結(jié)果不符合規(guī)定或供試品不適于用光阻法測定時,應(yīng)采用顯微計數(shù)法進行測定,并以顯微計數(shù)法的測定結(jié)果作為判定標準。”美國藥典、日本藥典、歐洲藥典也提到“供試品不適于光阻法時可以采用顯微計數(shù)法”,但并未出現(xiàn)“以顯微計數(shù)法的測定結(jié)果作為判定標準”這一表述。在美、日、歐藥典中,當出現(xiàn)光阻法和顯微計數(shù)法都可采用的情況時,并未明確說明以哪一個方法為準,而是希望得到一個一致(Conformance)的結(jié)論。
2.2.2 限定標準比較
在大體積和小體積的分類中,中國藥典、日本藥典將100 mL規(guī)定為大體積,美國藥典和歐洲藥典則將100 mL規(guī)定為小體積,除此之外,各藥典對于大容量注射劑和小容量注射劑的限定標準一致,具體見表6。
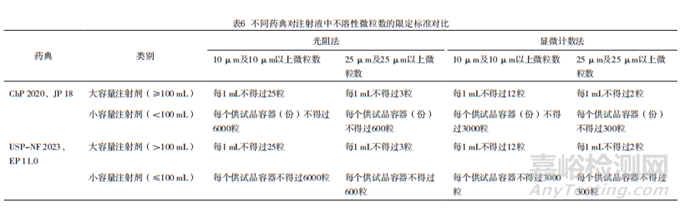
但是在ICH Q4B附錄3(R1)中,對于100 mL注射劑,歐盟和FDA均認為美、日、歐三方藥典的判定標準均有同等效力。因此,中、美、日、歐在對注射液中不溶性微粒數(shù)的限定標準方面要求一致。
2.3 系統(tǒng)適用性檢測的對比
系統(tǒng)適用性是在檢測樣品之前,用來確認測量系統(tǒng)的重復(fù)性和準確性是否能夠滿足當前分析的要求,測試基于的原則:整個測量系統(tǒng)是由儀器、電路、分析方法和樣品所組成的,將其作為整體去測試,從而驗證整個系統(tǒng)的狀態(tài)。藥典中對不溶性微粒檢測的系統(tǒng)適用性描述不涉及分析方法和樣品,只對儀器進行系統(tǒng)適用性要求(使用標準樣品對儀器進行校驗),因此本節(jié)對藥典中系統(tǒng)適用性的對比只涉及了儀器部分。
中、美、日藥典均介紹了不溶性微粒測試方法的系統(tǒng)適用性標準,歐洲藥典僅介紹了不溶性微粒測試方法,未見系統(tǒng)適用性要求相關(guān)內(nèi)容。中國藥典和日本藥典將測試方法、限定標準、系統(tǒng)適用性都放在同一章節(jié),分別對應(yīng)2020年版《中華人民共和國藥典》通則<0903>和JP 18<6.07>,USP-NF 2023則將測試方法和限定標準放在<788>中,而系統(tǒng)適用性測試標準則在<1788>、<1788.1>、<1788.2>中均有描述。
2.3.1 光阻法
歐洲藥典并未對光阻法的系統(tǒng)適用性進行較多描述,中國藥典、美國藥典、日本藥典在系統(tǒng)適用性檢測上的不同之處主要為檢測項目的差異,詳見表7。

(1)在儀器的校準頻次方面,中國藥典明確至少每6個月校準一次,日本藥典明確至少每一年校準一次,美國藥典雖未明確校準頻次,但是提出應(yīng)在使用前或日常進行足夠頻次的性能確認,以驗證儀器在制造商的規(guī)格和預(yù)期用途范圍內(nèi)提供準確、一致的結(jié)果。
(2)在體積準確性的驗證方面,美國藥典僅驗證2次結(jié)果的準確性,未考慮此項重復(fù)性的驗證。中國藥典則驗證3次測試準確性,其對體積準確性的要求更高。
(3)相對于中國藥典,美國藥典和日本藥典對流速和直徑校準提出了要求,檢測項更為詳細及嚴苛。事實上,基于光阻法原理,將流速作為系統(tǒng)適用性確認項有利于測試過程更準確。而且對于光阻類傳感器而言,不同流速下的校準曲線結(jié)果是有差異的。對于用于曲線校準的粒子的大小,美國藥典和日本藥典也有所不同,美國藥典中對于標準粒子的選擇進行了說明,比如10μm和25μm是標準檢測要求,增加5μm和15μm的校準是為了確保測試結(jié)果準確可靠。
(4)中、美、日藥典在對微粒計數(shù)的測定方法與標準上均有不同。中國藥典要求計數(shù)誤差在20%以內(nèi),而美國藥典要求計數(shù)誤差在10%左右,計數(shù)準確性要求更高,且針對不同類型的設(shè)備,作了3種方法區(qū)分,充分考慮不同設(shè)備的驗證能力。且在實踐操作中,美國USP官方商城中出售的Particle Count Set中將計數(shù)準確性設(shè)定了兩個范圍值,一個是顆粒數(shù)量的范圍值,另一個是粒徑15μm和10μm的顆粒數(shù)量比值,且經(jīng)過計算,誤差范圍約在±10%。
(5)在對傳感器分辨率的要求上,中國藥典和美國藥典對粒子大小分辨率測試方法的描述較為全面,日本藥典和美國藥典的檢查方法和要求一致,但計算方法略有不同。對比而言,美國藥典所述的計算公式更科學,更能反饋傳感器的分辨率性能。
2.3.2 顯微計數(shù)法
對于顯微計數(shù)法的系統(tǒng)適用性檢測,日、歐藥典內(nèi)容一致,且所涉及的項目在美國藥典均有包含。美國藥典所含內(nèi)容最為詳實,中國藥典在項目要求上相對較少,詳見表8。

美國藥典認為顯微鏡的光學對準和照片是顯微計數(shù)法成功的關(guān)鍵,且顯微鏡對準不良也會造成操作者的疲勞。由于衍射極限約為1μm,在次優(yōu)照明、干擾背景、粒子特征等多因素影響下,可能很難區(qū)分9μm和10μm或24μm和25μm。而操作員必須確認一個粒子是否高于或低于極限,因此優(yōu)化檢測系統(tǒng)的分辨率十分重要,基于操作者和保證測定數(shù)據(jù)的準確性考慮,美國藥典對顯微鏡、照明器、閾值靈敏度、微粒計數(shù)和直徑校驗(機器自動計數(shù)儀器)、微粒計數(shù)和直徑校驗(手動計數(shù)儀器)均作了詳細的要求。且美國藥典要求,當該粒子粒徑結(jié)果處于極限,不確定是否應(yīng)當被計數(shù)時,操作員應(yīng)當將其計數(shù),且應(yīng)對主要的粒子形態(tài)進行描述,適時做進一步分析。
在對微孔濾膜的要求上,中國藥典明確了孔徑為0.45μm,美國藥典則是給出了孔徑范圍(孔徑為1.0μm或更細),給予操作者一定的選擇空間,且在<1788.2>章節(jié)中對此進行了說明,即“更細的孔徑膜有更光滑的表面,提高了顆粒在顯微鏡下的分辨率,但較小的孔徑可能會阻礙實驗過程中黏性樣品液的過濾”。
3、對于生物技術(shù)藥物中不溶性微粒的監(jiān)管考慮
3.1 藥典要求
目前,ChP 2020對于不溶性微粒的要求僅有通則0903,通過對ChP 2020三部收載的治療性蛋白品種進行匯總,發(fā)現(xiàn)此類品種的不溶性微粒限度也僅要求符合通則0903,未見針對生物技術(shù)藥物特性提出不溶性微粒要求。而USP-NF 2023、EP 11.0、JP 18則均收載了生物技術(shù)藥物中不溶性微粒相關(guān)的檢測方法和要求。如USP-NF 2023和JP 18針對該類產(chǎn)品的特性,為治療性蛋白注射液不溶性微粒單獨制定了通則;EP 11.0雖未單設(shè)章節(jié),但相較于上一版,在內(nèi)容中也增加了生物技術(shù)藥物適用的部分,具體內(nèi)容如下:
(1)在檢測方法方面,USP-NF 2023<787>章提到應(yīng)首選光阻法,但同時也提到在充分研究的基礎(chǔ)上,其他監(jiān)管可接受的方法及限度要求也是可以被接受的。顯微計數(shù)法則在非蛋白粒子或重要的粒子特征檢查時會用到。在JP 18<6.17>章節(jié)中則明確檢查方法為光阻法,并未提及顯微計數(shù)法。EP 11.0<2.9.19>則提到光阻法和顯微計數(shù)法均可適用。另外,USP-NF 2023、EP 11.0、JP 18均提出,應(yīng)用光阻法檢測時,超聲處理作為常用的除氣泡的方式并不適用,因為超聲可能會導(dǎo)致蛋白的聚集或變性而影響檢測結(jié)果。
(2)在檢測用樣品方面,USP-NF 2023<787>章節(jié)指出,若企業(yè)全面考慮到使用較小的檢測產(chǎn)品體積可能涉及的分析相關(guān)的問題,在提供相關(guān)說明的情況下,允許使用較小的檢測產(chǎn)品體積和較小的檢測等分來進行檢測,單次檢測所需的體積通常為0.2~5.0 mL。在JP 18<6.17>中也明確樣品檢測體積可以減低為1 mL至5 mL,在滿足一定條件下,甚至可以降至0.2 mL。而EP 11.0<2.9.19>僅說明在儀器適用的情況下,可將檢測體積定為1 mL至5 mL。USP-NF 2023、EP 11.0、JP 18均認可若可以從一個容器中獲得測試所需的體積,則對單個容器進行測試;若單個容器中的體積不能滿足測試需求,可將多個容器中的內(nèi)容物混合;但均未對混合樣品的數(shù)量進行規(guī)定。與表2要求相比,這充分考慮到該類產(chǎn)品的容量較小的規(guī)格特征[18]和檢測成本。
(3)在限度要求上,均仍僅對粒徑為10μm和25μm及以上的粒子進行計數(shù),JP 18<6.17>和EP 11.0<2.9.19>中的限度要求與表6一致。USP-NF2023<787>雖對治療性蛋白注射劑中不溶性微粒提出了要求,但也僅在通用章節(jié)<788>要求的基礎(chǔ)上,對于大容量注射劑(>100 mL)增加了單個供試品容器總微粒負載量的要求,即對于粒徑在10μm及10μm以上微粒,數(shù)量應(yīng)≤6000粒/容器;對于25μm及25μm以上微粒,數(shù)量應(yīng)≤600粒/容器;在<1787>章節(jié)中提供了對產(chǎn)品中2~10μm粒徑粒子計數(shù)和區(qū)分粒子類型的檢測方法,以便在進行此類產(chǎn)品不溶性微粒檢測時可以進行更小粒徑范圍的粒子監(jiān)測。
但是,對有免疫原風險的小粒徑(0.1~10μm)不溶性蛋白聚集體微粒,均未提出明確要求或指導(dǎo)性建議以控制其可能帶來的免疫原性危害。
3.2 指導(dǎo)原則等相關(guān)要求
FDA在2014年發(fā)布的《行業(yè)指南:治療性蛋白制品免疫原性的評估》(Guidance for IndustryImmunogenicity Assessment for Therapeutic Protein Products)指出,蛋白質(zhì)聚集形成的顆粒可能會引起或增強免疫反應(yīng)[19],形成的高分子聚合蛋白是可能造成免疫反應(yīng)的主要物質(zhì)。此外,0.1~10μm大小范圍內(nèi)的微粒也具有很強的免疫原性潛力。因此,F(xiàn)DA建議企業(yè)應(yīng)在研發(fā)過程中盡可能采取措施減少蛋白質(zhì)聚集,在產(chǎn)品檢測中也需要采用單獨或聯(lián)合增強蛋白聚集體檢測的方法來表征產(chǎn)品中不同種類的聚合物。FDA在該文件中還指出,企業(yè)應(yīng)評估治療性蛋白產(chǎn)品在整個保質(zhì)期過程中(包括最初樣品)存在的亞可見顆粒(2~10μm)的范圍和水平。隨著科技的發(fā)展和更多檢測方法的出現(xiàn),企業(yè)應(yīng)努力在更小的尺寸范圍內(nèi)(0.1~2μm)對產(chǎn)品中的粒子進行表征。根據(jù)USP-NF 2023<1787>的描述,這里應(yīng)當包括粒子的類型、粒徑范圍及不同粒徑粒子的數(shù)量水平等內(nèi)容。
經(jīng)查詢?nèi)毡舅幤丰t(yī)療器械管理局(Pharmaceuticals and Medical Devices Agency,PMDA)、歐洲藥品管理局(The European Medicines Agency,EMA)和我國國家藥品監(jiān)督管理局相關(guān)官網(wǎng),PMDA于2023年3月在官網(wǎng)發(fā)布草案《生物技術(shù)產(chǎn)品(生物制藥)原料藥、藥品中不溶性顆粒物的流動成像法評價方法》[Evaluation Method of Insoluble Particulate Matter in Biotechnological Products (Biopharmaceuticals) Drug Substances/Drug Products by Flow Imaging Method〈G3-17-182〉],該草案提及了對于蛋白聚集體帶來的免疫原性,應(yīng)對其進行嚴格的評估和控制,但是該方法的測量范圍仍為2~100μm。國家藥品監(jiān)督管理局藥品審評中心于2022年5月發(fā)布的《特異性人免疫球蛋白藥學研究與評價技術(shù)指導(dǎo)原則》對于不溶性微粒控制也僅提出“特免制品尤其是靜注特免制品,還需重點關(guān)注對不溶性微粒的控制。”對于蛋白聚集體僅提出“特免制品制備過程中產(chǎn)生的多聚體與抗補體活性(ACA)有關(guān),應(yīng)重點控制免疫球蛋白G(IgG)多聚體含量。”未見對于多聚體的含量和小粒徑不溶性蛋白聚集體顆粒相關(guān)標準和指導(dǎo)原則。
4、建議
對于治療性蛋白注射液中不溶性微粒,雖然FDA發(fā)布行業(yè)指南建議評估治療性蛋白產(chǎn)品中小粒徑(0.1~10μm)的不溶性微粒,但是該行業(yè)指南并非強制要求。所以,對于產(chǎn)品中不溶性微粒,目前主要依靠企業(yè)的風險防控手段進行控制,企業(yè)在進行充分的研究和有足夠的數(shù)據(jù)支持的基礎(chǔ)上,明確在新增的不溶性微粒不影響產(chǎn)品本身質(zhì)量的前提下,采取終端過濾的方式除去;如抗EGFR全人源單抗注射液,其進口原研藥(帕尼單抗)和國內(nèi)生物類似藥注射前都要進行濾器過濾。可是,這并未從根本上解決問題,還是需要研發(fā)者秉著質(zhì)量源于設(shè)計(QbD)的理念在藥物的研發(fā)設(shè)計時,關(guān)注到產(chǎn)品中的小粒徑不溶性微粒,通過制定有效的措施,減少不溶性微粒的產(chǎn)生以降低或消除風險。
小粒徑(0.1~10μm)蛋白聚集體通過靜脈注射進入患者體內(nèi)可能引發(fā)的免疫原性的風險日益受到關(guān)注,可是目前仍按照藥典通則要求對10μm及10μm以上的粒子進行質(zhì)量控制。為更好地控制生物技術(shù)藥物中小粒徑(0.1~10μm)蛋白聚集體可能帶來的免疫原性風險,保障患者的用藥安全,提出以下三點建議:
(1)方法學研究方面:建議應(yīng)使用多種方法檢測注射液中小粒徑不溶性微粒,進行全面的表征和對比性研究以獲得準確的數(shù)據(jù)。此外,產(chǎn)品中“固有顆粒”的形成是一個動態(tài)、持續(xù)的過程,建議可分兩個階段對產(chǎn)品中小粒徑微粒進行監(jiān)測。先建立快速檢測方法,準確識別產(chǎn)品中小粒徑(0.1~10μm)不溶性微粒的增長情況;再根據(jù)需要對產(chǎn)品中不溶性微粒進行鑒定和分類,以充分了解產(chǎn)品的質(zhì)量情況。
(2)數(shù)據(jù)收集方面:建議持續(xù)收集產(chǎn)品中小粒徑不溶性微粒數(shù)據(jù),包括臨床試驗樣品、穩(wěn)定性考察樣品及上市后的貨架期內(nèi)的樣品,結(jié)合不良反應(yīng)情況對數(shù)據(jù)進行分析。
(3)技術(shù)要求方面:建議在可能涉及此類風險的生物技術(shù)藥物藥學研究指導(dǎo)原則中明確對初始樣品和穩(wěn)定性樣品中0.1~10μm小粒徑不溶性微粒數(shù)據(jù)的收集,以引導(dǎo)企業(yè)關(guān)注,盡可能在研發(fā)階段找到合理的措施降低風險。
編者按:
綜上所述,不同藥典中不溶性微粒檢測方法經(jīng)歷了不斷的發(fā)展和完善。未來,隨著科技的進步和醫(yī)藥行業(yè)的發(fā)展,不溶性微粒檢測將面臨新的挑戰(zhàn)和機遇。我們需要繼續(xù)加強研究和合作,不斷完善和優(yōu)化不溶性微粒檢測方法,以確保注射劑的安全性和有效性。

來源:《中國藥事》


