您當前的位置:檢測資訊 > 科研開發
嘉峪檢測網 2020-09-07 10:17
IND、 NDA、BLA、ANDA與OTC,是我們談到一個新藥時經常聽到的詞。它們分別代表什么?具體有什么區別?了解一下FDA整個新藥的批準流程會讓你更好理解這幾個詞的意義。
FDA的新藥審評包括了兩個過程:一個是新藥臨床試驗申請(簡稱IND)審評過程,另一個是新藥上市申請(簡稱NDA)審評過程。
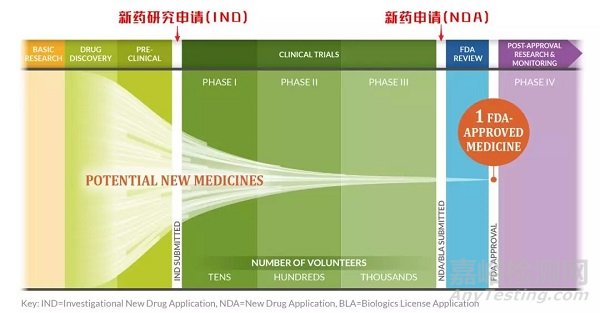
新藥申請的步驟
名詞短語:
IND:新藥臨床試驗申請(Investigational New Drug)
NDA:新藥生產上市注冊申請(New Drug Application )
BLA:生物制品許可申請(Biologics License Application)
ANDA:仿制藥注冊申請(Abbreviated New Drug Application)
OTC:非處方藥(Over The Counter)
FDA藥品批準程序
FDA藥品批準程序可大致分為以下幾個步驟:
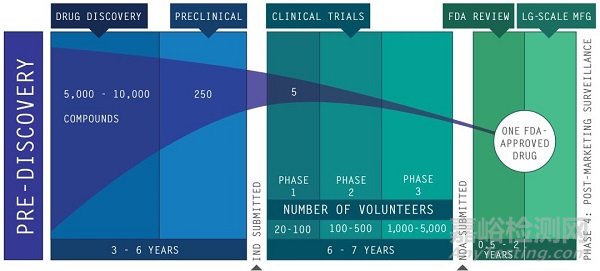
1.臨床前試驗(Pre Clinical)
研究人員將前期篩選出的新化合物進行動物試驗,證明發現的新化合物對某種疾病具有生物活性,同時還會評估該化合物的安全性。
臨床前研究用來評估:
(1)藥品的藥理學現象和作用機理(MOA);
(2)藥物毒性特征和毒性靶器官;
(3)藥物吸收、分布、代謝和排泄(ADME)。當藥品申辦者認為它已具有足夠的數據證明該藥是安全時,就可準備向FDA提交新藥臨床研究申請(IND)。
2.新藥臨床試驗申請(IND)
化合物通過臨床前試驗后,需向FDA提交IND申請,以便可以將該化合物應用于人體試驗。
詳解新藥臨床試驗申請(IND)
IND的英文全稱是Investigational New Drug,中文被稱為:新藥臨床試驗申請。就是說當一個新藥當決定進入臨床試驗時,則要向FDA提交IND(新藥研究的申請),同時報送所有研究資料。FDA在收到IND以后,在一個月內必須給予答復。如果申請人在一個月內得不到FDA的答復,即表示已經批準進入人體試驗,可自動進入臨床研究。
美國法律規定,藥品在被運輸到其他國家時應是獲得上市許可批準的。由于申辦者可能會把在研藥物交給其他國家的臨床研究者,因此申辦者必須尋求法律上可以豁免的一種方式。IND就是申辦者嚴格根據法律意義在FDA獲得豁免權的方式。
IND主要目的
提供足夠信息來證明藥品在人體進行試驗是安全的,以及證明針對研究目的的臨床方案設計是合理的。在IND申報階段,FDA一般規定(最低限度)藥品申辦者必須:
(1)做該藥的藥理研究;
(2)在至少2種動物身上進行急毒試驗;
(3)按照該藥預想的用途進行為期2個星期至3個月的短期研究。一旦臨床前研究結束,動物試驗并沒有隨之完成,許多時間更長、更專項的研究如慢性、抗癌試驗將在整個新藥申請過程中進行。
3.臨床試驗(Clinical Trials)
臨床試驗(Clinical Trials)指任何在人體進行的藥物的系統性研究,以證實或揭示試驗藥物的作用、不良反應及/或試驗藥物的吸收、分布、代謝和排泄,目的是確定試驗藥物的療效與安全性, 臨床試驗一般分為I、II、III、IV期臨床試驗。
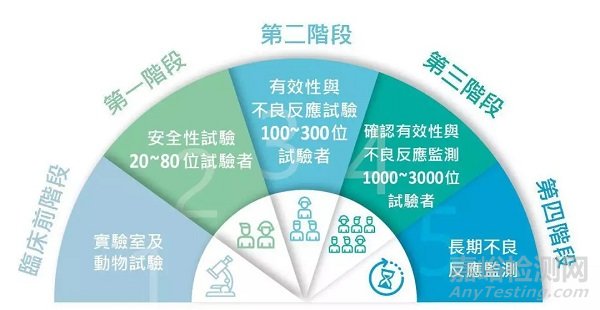
Ⅰ期:臨床藥理學及人體安全性評價。受試者為健康成年志愿者。
Ⅱ期:治療作用初步評價階段。受試者必須是患者。劑量探索階段稱為Ⅱa期,評估藥物有效階段稱Ⅱb期。
Ⅲ期:治療作用確證階段。進一步驗證藥物對目標適應癥患者的治療作用和安全性,評價利益與風險關系,為藥物申請的審查提供充分依據。
Ⅳ期:新藥上市后應用研究階段。考察在廣泛使用條件下藥物療效和不良反應;評價在普通或者特殊人群中使用的利益與風險關系及改進給藥劑量等。
4.NDA或BLA申請
在臨床試驗結束后,申請者向FDA提交NDA或BLA,申請批準上市。審評過程中如臨床試驗結果不詳盡,FDA會要求補充試驗以證明藥物安全性和有效性。
生物制品許可申請(BLA)
BLA(生物制品許可申請) 是Biologic License Application的簡稱,是向美國FDA提交用于支持評審和最終批準生物制品在美國上市和銷售的文件材料。
生物制品的上市銷售申請是根據公共衛生醫療服務(PHS)法案中的有關條款進行批準的。該法案要求生產生物制品的公司在跨州進行產品銷售時需要持有相關的產品許可證。
BLA指的是一個包含有生物制品的生產工藝、化學、藥理學、臨床藥理學和醫學影響方面特定信息的遞交材料。如果提供的材料符合FDA的要求,那么申請便會得到批準并頒發給生產企業相關產品上市銷售的許可證。
新藥申請(NDA)
新藥申請(New Drug Application),當申辦者有足夠理由能證明藥品的安全性和有效性滿足了FDA對于上市的要求時,申辦者就可以向FDA遞交NDA了。所有的新藥要在美國上市必須經過新藥評審過程。遞交材料中必須包含如化學、藥理、生物藥劑和統計學等方面的技術材料供評審。
如果NDA獲得批準,那么藥品就能在美國上市了。此外為了便于內部追蹤,所有的NDA申請都會有一個NDA號。一般NDA的評審與IND的評審是由同一個評審組評審的,但是NDA無疑會更為耗時。
植物藥NDA申報材料與化學藥品類似,主要包括以下內容:CMC數據、非臨床藥理和毒理數據、人體藥代動力學和生物利用度數據、微生物數據、臨床數據、安全性數據更新報告、統計學數據、病例報告表、有關專利情況、樣品、包裝及標簽等。
一般符合以下情況均可向FDA提出NDA申請:
(1)新分子實體(NME);
(2)新化學實體(NCE);
(3)原批準藥品相同化學成分的新鹽基、新酯基;
(4)原批準藥品的新配方組成;
(5)原批準藥品的新適應癥(包括處方藥轉非處方藥使用);
(6)新劑型、新給藥途徑、新規格(單位含量);
(7)兩種以上原批準藥品的新組合。
這里需要需要提一下FDA四條加快創新藥品上市的特殊審批通道,其主要包括:突破性療法(Breakthrough)、優先審評(Priority review)、快速通道(Fast track)、加速批準(Accelerated approval)。
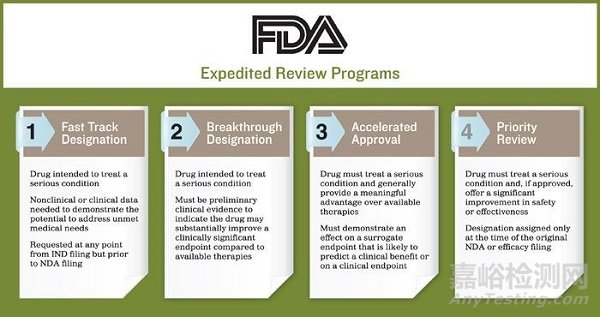
5.批準上市(FDA Approval)
獲批后,FDA會在網站公布藥物批準類型及藥物說明書。
仿制藥注冊申請
仿制藥注冊申請ANDA的英文全稱為Abbreviated New Drug Application,中文稱為仿制藥注冊申請。
仿制藥需要在原料藥、劑型、規格、給藥途徑和使用條件(除非因專利問題無法做到使用條件相同)等方面與已上市藥品相同。不僅如此,仿制藥還需要跟原研藥在質量、作用和適應癥( intended use )上與原研藥一致,故又被稱為通用名藥或非專利藥。
他們在FDA的申請被稱為“ Abbreviated New Drug Application ”,是因為所仿制的原研藥生產商已經做了動物和人體的相關實驗并證明了藥物的安全性和有效性,因此仿制藥申請不需要重復這些實驗,只需要科學地證明仿制的藥品與原研是生物等效的即可。
(1)仿制藥的基本要求:
仿制藥的基本要求是要實現仿制藥與原研藥的治療等效( Therapeutical Equivalence ), 而仿制藥要實現與原研藥的治療性等效,不僅需要與原研藥藥學等效,
還需要與原研藥能夠生物等效。
主要包括:
a. 活性成分必須與原研藥相同。
b. 劑型與原研藥相同。(若原研藥是片劑,那么仿制藥也必須是片劑,不能做出膠囊或其他劑型。FDA甚至還要求仿制藥的外形與外觀,與原研藥要保持近似但又不能夠完全雷同。)
c. 規格與原研藥相同(如原研藥是30mg,那么仿制藥品也必須是30mg)。
d. 服用方式相同,例如原研藥是口服,那么仿制藥也必須是口服。
e. 使用條件需要相同。
f. 仿制藥的強度、純度、質量等方面與原研藥相同。
g. 仿制藥的生產條件需要原研藥相同。
對于一般口服制劑來說, FDA不要求仿制藥的輔料(一些特定的關鍵輔料除外)與原研藥一樣,但是對于注射劑來說,仿制藥的輔料(緩沖液、防腐劑、抗氧化劑除外)是需要與原研藥保持一致的。
(2)生物等效(BE,Bioequivalence)
生物等效(BE,Bioequivalence) 是指仿制藥和原研藥到達人體的時間和速度是相當的。通常證明生物等效的一個常見的方法是測定仿制藥到達健康人體( 24-36 名)血液所需的時間和血液濃度,如果仿制藥能夠在同樣的時間內將同樣量的活性成分傳輸到人體的血液中,則視為二者等效。
BE試驗是為了比較仿制藥和原研藥體內吸收程度和吸收速率,因此提供源于BE試驗的數據是ANDA申報資料中一個關鍵的組成部分。與評估藥學等效性一起,確立生物等效性允許在藥政監管層面上做出治療等效性的結論。
非處方藥(OTC)
OTC是over the counter的縮寫,即非處方藥,是指那些不需要醫生處方,消費者可直接在藥房或藥店中即可購取的藥物。
非處方藥是由處方藥轉變而來,是經過長期應用、確認有療效、質量穩定、非醫療專業人員也能安全使用的藥物。
對每類非處方藥,FDA都制定了一個專論,凡是符合專論的藥品,可以直接作為非處方藥上市而不必得到FDA的批準。但如不符合專論,在必須單獨經過新藥申請評審過程,但這一過程主要用于新的活性成分第一次作為非處方藥的情況。

來源:銘研醫藥


