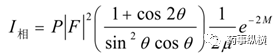前言:行業內公認,X-射線衍射圖譜(XRD)是確定多晶型最可靠的依據,XRD圖譜也常被稱為晶型的“指紋”。由于有機藥物晶體峰位多且雜,重疊嚴重,嚴格意義上的單純圖譜比較,往往得出生產藥品晶型與標準品完全不同的結論,這種圖譜對比差異在原料藥申報及研究生產過程中帶來諸多困擾。因此需要根據研究目的,有策略的系統分析XRD圖譜數據。
XRD圖譜分析中的主要問題
1、粉末衍射XRD圖譜與標準圖譜比對
涉及到晶型的原料藥申報和研究生產中,均需要與標準品或者理論圖譜比較,粉末X-射線衍射儀測試后處理得到的XRD數據分為以下三類:(標準圖譜的獲取方式詳見20201215期推文:《被忽略的“XRD標準譜”的獲取技巧》)。
(1)2θ角度和晶面間距(d值)確定多晶型類型,是藥物API中原子和分子的排列方式及位置信息;是晶型的特征信息。
(2)峰高度(Hight)、峰強度(Int.I)、相對峰強度(Rel.int.)是晶型中不同晶面分布的統計,是樣品晶型的特征信息,也是晶型定量分析的基礎。
(3)半峰寬(FWHM)表征衍射峰的峰形,反映多晶體晶粒尺寸、外形、尺寸分布等性質,不作為晶型的特征信息。
藥物晶型研究中,最常用的是通過2θ(或d-spacing)與相對峰強度Rel.Int比較晶型的一致性。但由于峰強度受樣品制備、測試條件、儀器等影響較大,對于原料藥申報等重要圖譜比對和晶型定量分析中,則需要單獨開發樣品制備、測試等方法,保證樣品峰位置和峰強度的可重現性。對于需要兼顧效率的批次圖譜的對比:如專利晶型確定、晶型篩選等,大家公認:(1)粉末XRD圖譜的峰位置(或者晶面間距d值)有時比強度更重要;(2)藥物晶體中低角度的數據比高角度數據更重要;(3)有機藥物晶體在40°以上基本無鑒別意義的衍射峰(晶面間的距離越大,角度越小,晶面上的原子排列越密集)。
2、峰缺失和衍射峰強度次序變化
晶型相關研究及實踐過程中,經常會遇到PXRD圖譜少量峰缺失,峰強度不一致的情形,對于遇到這類問題,常結合熱分析DSC/TGA、固體紅外、拉曼等輔助數據判斷其是否為新晶型,同一晶型出現峰的缺失和衍射峰強度次序變化有以下幾種可能原因:
(1)“晶體粒度”的影響
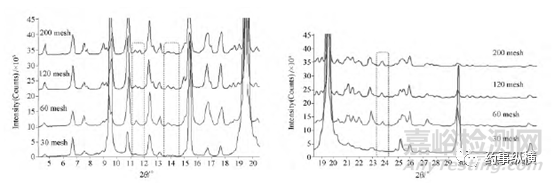
利福平晶型II樣品,依次經過30、60、120、200目的篩子后測試粉末衍射圖譜,發現同一樣品,過篩為不同粒度級別的樣品進行測試,得到的XRD圖譜峰強度及強度次序發生明顯變化,并且局部放大圖顯示,過30目篩子(0.6mm)的譜圖,存在明顯的“峰缺失”(圖2框線標注部分),如下圖1/2所示:
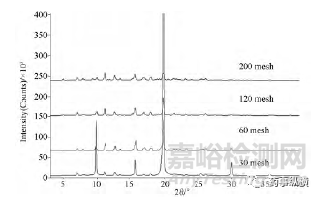
圖1利福平同一樣品過不同篩目后,XRD圖譜峰強度差異巨大
圖2利福平同一樣品過不同篩目后,XRD圖譜局部放大圖顯示部分峰的缺失示意圖
晶型粒度對“峰缺失”和峰強度影響原因:衍射強度本質是對不同晶面分布的統計。因此XRD測試中,理論上1cm2的面積上要有超過1100萬個衍射數據才具有統計的意義。我們常用的X-射線粉末衍射儀,測試的樣品對象是“一堆多晶粉末”,且其中的每一粒粉末都是“多晶體”。“多晶體”是由多個小晶體組成的晶體結構。每個小晶體(即單晶)的內部,晶格位向是均勻一致的,而各個小晶體之間,彼此的位向卻不相同。粒度較大的“多晶”樣品,內部小晶體間的各項異性導致不同粒度產品峰強度差異顯著,甚至出現少量峰缺失(即大家熟知的“擇優取向”)。因此X-射線粉末衍射樣品制備中,樣品需要經過研磨處理,一般情況下,樣品手感像面粉,有滑膩的感即可。而對于晶型含量的定性研究中,研磨樣品需要過360目篩的粉末顆粒用于分析。
(2)制樣過程“研磨”的影響
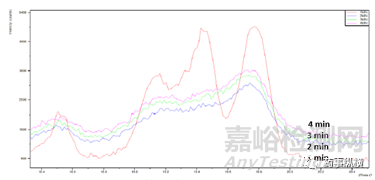
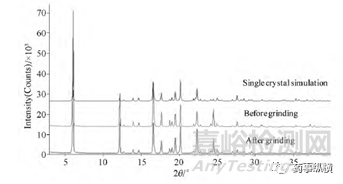
如前所述,通常對樣品進行研磨降低大粒度樣品“擇優取向”的影響。但“研磨”有導致晶型轉變(壓力轉晶)和結晶度降低的風險,從而導致不同程度的“峰缺失”或“峰強度”改變。如某一致性評價藥品晶體,研磨2min、3min、4min的樣品與研磨1min的樣品對比,19.4°處(特征強峰)的峰缺失。又如圖4布洛芬晶體,研磨前后,峰強度差異明顯。
圖3某一藥物晶體不同研磨時間的XRD放大圖譜
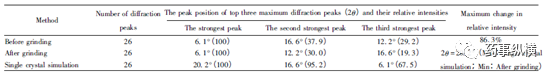
圖4布洛芬研磨前后以及單晶模擬的XRD圖及差異圖表
(3)晶體形貌的影響
對于“薄片狀”和“細長針狀”形貌的晶體,特點是長徑比非常大。采用壓片法制樣,幾乎無法消除擇優取向的影響。測試得到的圖譜特定晶面峰缺失嚴重,峰強度重復性差。特殊裝樣方法:如撒樣或側法裝樣,是這類特殊晶體形貌樣品得到可重復圖譜的關鍵。
(4)樣品試片平面的影響
試片表面形狀不規則、毛糙、向上或向下凹凸等,會引起XRD衍射圖譜峰的寬化、峰位置的位移、衍射峰強度重現性差。制樣過程中,要求輕輕壓片磨平。
此外,粉末XRD的峰位置、相對峰強度受洛倫茲極化、吸收、溫度等的影響,衍射儀法的衍射峰相對強度公式如下:
在對晶型XRD強度允許的誤差范圍是5%;峰位置的偏差限度為±0.2°。因此對于定量分析及重要批次的圖譜重復性,在設備、制樣等一致情況下,滿足上述2θ±0.2°,峰強度5%限度要求。
3、峰形:半高寬、峰對稱性
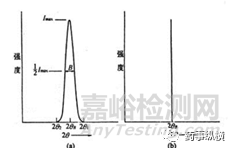
完美晶體(理想晶體)的衍射峰形為一條直線,而實際晶體衍射峰有一定的寬度;在衍射強度一半的高度對應的一個峰形的半寬高(B),如下圖5所示。
圖5 (a)實際晶體的衍射峰形;(b)理想晶體的衍射峰形
衍射峰的峰寬受三個因素的影響:儀器本身造成的固有寬度、晶格畸變引起的寬化和晶粒尺寸影響。這三部分對衍射峰峰形的影響并不是簡單的累加作用,而是一種“卷積”關系。晶體XRD圖譜的峰形不作為晶型的特征信息。
4、結晶度與無定形混晶的區分
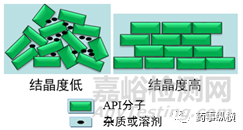
結晶度的概念來源于材料領域,是用來表示聚合物中結晶區域所占的比例。圖7藥物晶體領域結晶度的示意圖,藥物晶型結晶度類似表示晶體結晶區域的比例。即分子層面晶體的排列方式是一致的(晶型一致,因為晶型的定義即是分子的排列方式),但排列“緊密程度”存在區別,導致晶型結晶度的不一致。
圖6 藥物晶型結晶度示意圖
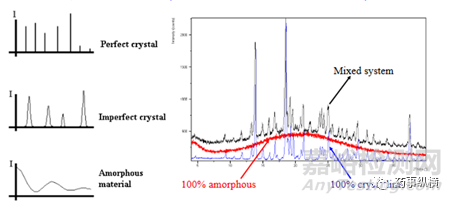
圖7 標示“mixed system”的XRD圖譜,可能是混有一定量的無定形,但也可能
是同一種晶型,只是結晶工藝的差異,得到的同一種晶型結晶度有差異。
圖7 結晶度or無定形混晶的XRD示意圖
5、2θ角度與晶面間距d相互轉換
晶型相關專利及文獻中,經常會隨機的只給出2θ角度值或者晶面間距d值,類似情況經常需要我們通過布拉格公式進行換算。2θ角度和晶面間距滿足拉格定律:2dsinθ=n*λ(d為平行原子平面的間距,λ為入射波波長,θ為入射光與晶面之夾角,n為衍射級數),即二者之間數據可以互換計算。X射線入射波長是通過濾波鏡的單色光(特定波長)即λ為定值:藥物晶體最常使用的cu靶X射線波長為1.5406;n為衍射級數,XRD衍射中,級數為1;根據布拉格定理,相互換算計算方式如圖所示:
圖8 2θ角度與晶面間距d相互轉換晶的計算方法
小結:藥物晶體X-射線衍射技術主要包括單晶X-射線衍射技術和粉末X射線衍射技術。單晶X射線衍射技術本身可以單獨完成結構、組分、含量、構型晶型等各類分析,它技術難度在于獲取可解析的單晶。而粉末X射線衍射圖譜,主要用于多晶形態的識別,測試結果影響因素較大,技術難點在于有策略的、系統的分析圖譜。
參考文獻:
[1]章中華, 徐銀榮,盛曉霞,等. 不同藥典指導原則下X射線衍射法檢測結果對藥物晶型判
定的影響[J]. 中國藥學雜志, 2017, 052(005):409-413.
[2] 江任之, 王玲. 特殊形貌原料藥晶型測定方法的研究[J]. 智慧健康, 2019, 5(10):41-43.
[3] 呂揚, 杜冠華. 晶型藥物[M]. 人民衛生出版社, 2009.
[4] 梁向暉, 鐘偉強, 毛秋平. X射線衍射儀的維護與使用[J].分析儀器, 2015, 000(005):89-91.
[5] 常穎, 鄭啟泰, 呂揚. X射線衍射分析技術在藥物研究中的應用[J]. 物理, 2007, 36(006):452-459.
[6] 徐勇, 范小紅. X射線衍射測試分析基礎教程[M].化學工業出版社, 2014.
[7] 胡恒亮, 穆祥祺. X射線衍射技術[M]. 紡織工業出版社, 1988.